1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
|
use strict;
use warnings;
use Bio::SeqIO;
my @gbk = glob("*.gbk");
foreach (@gbk) {
$_=~/(.+).gbk/;
my $str = $1;
open IN, "$_" || die;
local $/ = "LOCUS";
<IN>;
while (<IN>) {
chomp;
$_=~/\s+(\S+)/;
my $scaf = $1;
my $out = $str . "_" . $scaf . ".gb";
my $assession = $str . "_" . $scaf;
$_=~s/ACCESSION.+/ACCESSION $assession/g;
open OUT, ">$out" || die;
print OUT "LOCUS$_";
close OUT;
}
close IN;
}
$/ = "\n";
my @gb = glob("*.gb");
foreach (@gb) {
$_=~/(.+).gb/;
my $out = $1 . ".island";
system("islandpath $_ $out");
}
foreach my $gb (@gb) {
$gb=~/(.+).gb/;
my $str = $1;
my $out = $str . ".list";
my $seqin = Bio::SeqIO->new( -format => 'genbank', -file => "$gb");
open OUT, ">$out" || die;
while( (my $seq = $seqin->next_seq) ) {
foreach my $sf ( $seq->get_SeqFeatures ) {
if( $sf->primary_tag eq 'CDS' ) {
my @tags = $sf ->get_all_tags();
print OUT $sf->get_tag_values('locus_tag'), "\t", $sf->start, "\t", $sf->end, "\t", $sf->strand, "\t", $sf->get_tag_values('product'), "\t", $sf->get_tag_values('translation'),"\n";
}
}
}
}
my (%hash, %gi, %list, %gif);
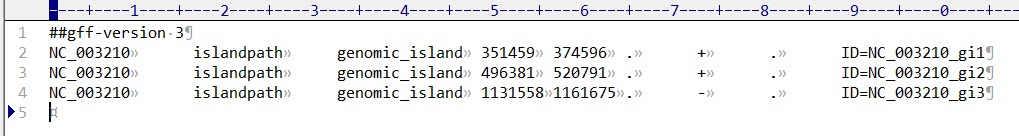
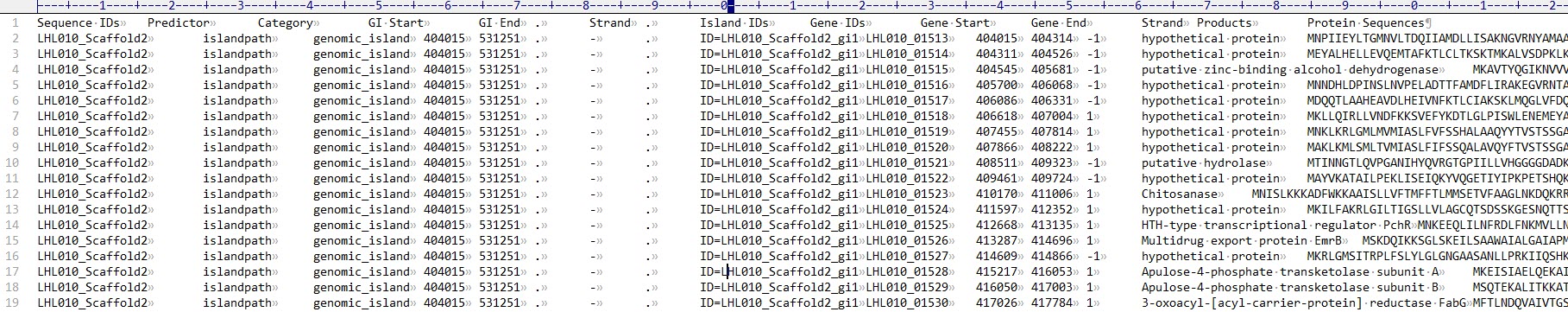
open OUT, ">All_island.txt" || die;
print OUT "Sequence IDs Predictor Category GI Start GI End . Strand . Island IDs Gene IDs Gene Start Gene End Strand Products Protein Sequences\n";
open LIST, ">All_island.list" || die;
print LIST "Island IDs\tGI Start\tGI End\tGI Length\tGene Number\tGene IDs\n";
my @GI = glob("*.island");
foreach (@GI) {
$_=~/(.+).island/;
my $list = $1 . ".list";
open IN, "$_" || die;
<IN>;
while (<IN>) {
chomp;
$_=~s/[\r\n]+//g;
my @lines = split /\t/;
my $start = $lines[3];
my $end = $lines[4];
my $id = $lines[-1];
my $gilen = $end - $start + 1;
$hash{$id} = $start . "-" . $end . "-" . $id;
$gi{$id} = $_;
$gif{$id} = $start . "\t" . $end . "\t" . $gilen;
}
close IN;
open GB, "$list" || die;
while (<GB>) {
chomp;
my @line = split /\t/;
foreach my $ids (sort keys %hash) {
my ($start2, $end2, $gi) = split /\-/, $hash{$ids};
if (($line[1] >= $start2) && ($line[2] <= $end2)) {
print OUT $gi{$gi} . "\t" . $_ . "\n";
push @{$list{$gi}}, $line[0];
}
}
}
close GB;
}
close OUT;
foreach (sort keys %list) {
print LIST $_ . "\t" . $gif{$_} . "\t" . @{$list{$_}} . "\t" . join (" ", @{$list{$_}}) . "\n";
}
close LIST;
|