
介绍
本文档描述了xcms(version>=3)的LCMS实验的数据导入、探索、预处理和分析。示例和基本工作流程改编自Colin A.Smith的原始LC/MS预处理和分析。
新版本使用XCMSnExpe对象(而不是旧的xcmsSet对象)作为预处理结果的容器。然而,为了支持依赖于xcmsSet对象的包和管道,可以使用as方法(即xset<-as(x, “xcmsSet”))将XCMSnExpe转换为xcmsSet对象,其中x是XCMSnxp对象。
安装
1 | if (!require("BiocManager", quietly = TRUE)) |
导入数据
xcms支持(AIA/ANDI)NetCDF、mzXML和mzML格式的LC/MS数据。对于实际数据导入,使用Bioconductor的mzR。出于演示目的,我们将分析[1]中的数据子集,其研究了敲除脂肪酸酰胺水解酶(FAAH)基因在小鼠中的代谢后果。原始数据文件(NetCDF格式)随faahKO数据包提供。数据集包括来自6只敲除小鼠和6只野生型小鼠脊髓的样本。每个文件包含在正离子模式下以200-600 m/z和2500-4500秒采集的质心模式(centroid mode)数据。为了加快处理速度,我们仅分析8个文件,且限定保留时间范围为2500到3500秒。
下面我们加载所有必需的包,在faahKO包中找到原始CDF文件,并构建描述实验设置的表型(phenodata)数据框。注意,对于实际实验,建议定义一个文件(表),其中包含原始数据文件的文件名以及每个文件的样本描述,作为附加列。然后,可以使用例如read.table导入该文件作为变量pd(而不是在下面的示例中在R中定义),并且可以将文件名传递给下面的readMSData函数,例如files=paste0(MZML_PATH, "/", pd$MZML_file),其中MZML_PATH是文件所在目录的路径,"mzMLfile"是phenodata文件中包含文件名那一列的列名。
1 | library(xcms) |
随后,我们使用MSnbase包中的readMSData方法将原始数据加载为OnDiskMSnExpe对象。MSnbase为质谱数据处理提供了基础结构和基础设施。此外,MSnbase可用于形心剖面模式(centroid profile-mode)MS数据(参见MSnbase包中的相应介绍)。
1 | raw_data <- readMSData(files = cdfs, pdata = new("NAnnotatedDataFrame", pd), mode = "onDisk") |
接下来,我们将数据集限制在2500到3500秒的保留时间范围内。这仅仅是为了减少该示例的处理时间。
1 | raw_data <- filterRt(raw_data, c(2500, 3500)) |
生成的OnDiskMSnExpe对象包含关于spectrum数量、保留时间、测量的总离子电流等的一般信息,但不包含完整的原始数据(即每个测量光谱的m/z和强度值)。因此,它的内存占用相当小,是代表大型代谢组学实验的理想对象,同时允许执行简单的质量控制、数据检查和探索以及数据取子集操作。m/z和强度值根据需要从原始数据文件中导入,因此原始数据文件的位置在初始数据导入后不应更改。
初始数据检查
OnDiskMSnExp按spectrum组织MS数据,并提供intensity、mz和rtime方法以访问文件中的原始数据(测量的强度值、相应的m/z和保留时间值)。此外,spectra方法可用于返回封装在spectrum对象中的所有数据。下面我们从对象中提取保留时间值。
1 | head(rtime(raw_data)) |
1 | ## F1.S0001 F1.S0002 F1.S0003 F1.S0004 F1.S0005 F1.S0006 |
即使实验由多个文件/样本组成,所有数据都会作为一维矢量(one-dimensional vectors,rtime的一个数值型向量,一个向量列表for mz和intensity,每个各自都包含一个spectrum的值)返回。fromFile函数返回一个整型向量,提供值到原始文件的映射。下面我们使用fromFile索引按文件组织mz值。
1 | mzs <- mz(raw_data) |
1 | ## [1] 8 |
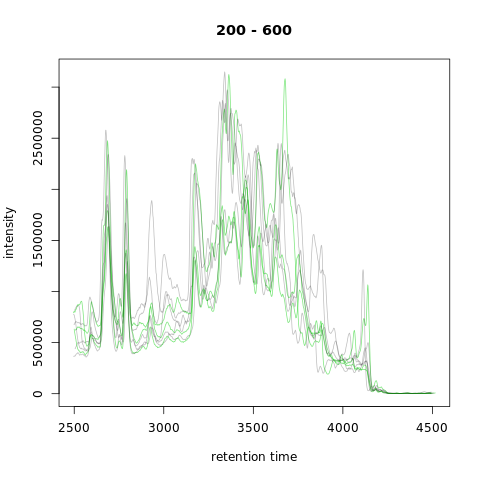
作为数据的第一次评估,我们在实验中绘制了每个文件的基峰色谱图(base peak chromatogram, BPC)。我们使用chromatogram方法并将aggregationFun设置为"max",以返回每个spectrum的最大强度,从而根据原始数据创建BPC。为了创建总离子色谱图(total ion chromatogram),我们可以将aggregationFun设置为sum。
1 | ## Get the base peak chromatograms. This reads data from the files. |
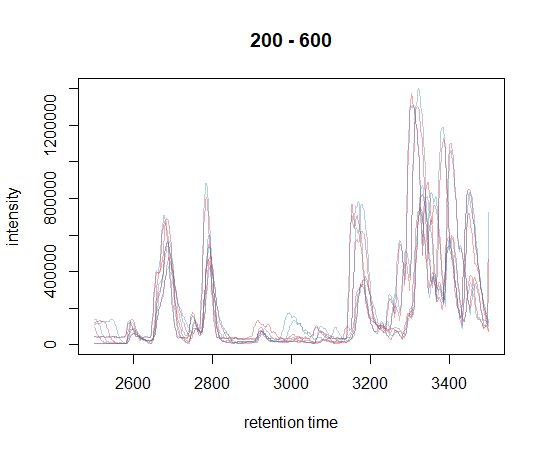
chromatogram返回了一个MCchromatograms对象,该对象以二维矩阵形式组织各个Chromatogram对象(实际上包含chromatographic数据):列表示样本,行(可选)表示m/z和/或保留时间范围。下面我们提取第一个样品的chromatogram,并获取其保留时间和强度值。
1 | bpi_1 <- bpis[1, 1] |
1 | ## F1.S0001 F1.S0002 F1.S0003 F1.S0004 F1.S0005 F1.S0006 |
1 | head(intensity(bpi_1)) |
1 | ## F1.S0001 F1.S0002 F1.S0003 F1.S0004 F1.S0005 F1.S0006 |
chromatogram法还支持从MS数据的m/z-rt切片中提取chromatographic数据。在下一节中,我们将使用此方法为选定的峰创建提取的离子色谱图(extracted ion chromatogram, EIC)。
注意,chromatogram从每个文件读取原始数据以计算chromatogram。另一方面,bpi和tic方法不从原始文件读取任何数据,而是使用输入文件的头定义(header definition)中提供的相应信息(可能与实际数据不同)。
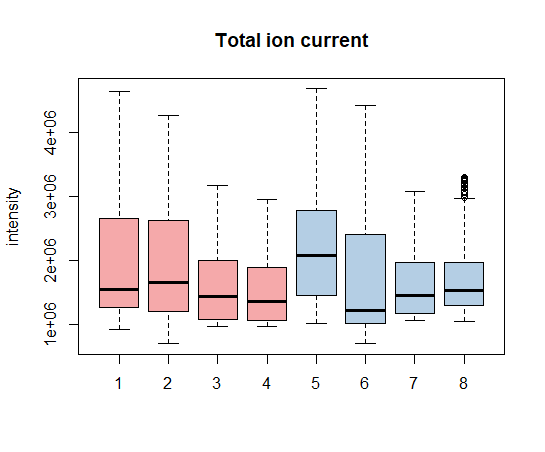
下面我们创建表示每个文件总离子流分布的箱线图。这些图对于发现有问题或失败的MS运行非常有用。
1 | ## Get the total ion current by file |
此外,我们可以根据样品的BPC的相似性对样品进行聚类。这也有助于在实验中发现潜在的有问题的样本,或获得实验中样本分组的初步概述。由于样本之间的保留时间不完全相同,我们使用bin函数将强度分组在沿保留时间轴的固定时间范围(bins)中。在本示例中,我们使用的bin大小为2秒,默认值为0.5秒。聚类是使用完全连锁分级聚类(complete linkage hierarchical clustering)对合并的BPC的成对相关性进行的。
1 | ## Bin the BPC |
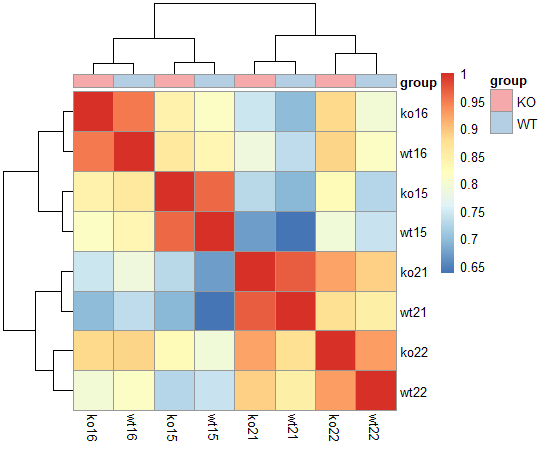
样本以成对方式聚类,样本索引的KO和WT样本具有最相似的BPC。
色谱峰检测
接下来,我们使用centWave算法进行色谱峰检测[2]。然而,在运行峰值检测之前,强烈建议目视检查内参或已知化合物的提取离子色谱图,以评估和调整峰值检测设置,因为默认设置不适用于大多数LCMS实验。centWave的两个最关键的参数是peakwidth(色谱峰宽度的预期范围)和ppm(对应于一个色谱峰的centroids m/z值的最大预期偏差;这通常比制造商规定的ppm大得多)参数。为了评估典型的色谱峰宽度,我们绘制了一个峰的EIC。
1 | ## Define the rt and m/z range of the peak area |
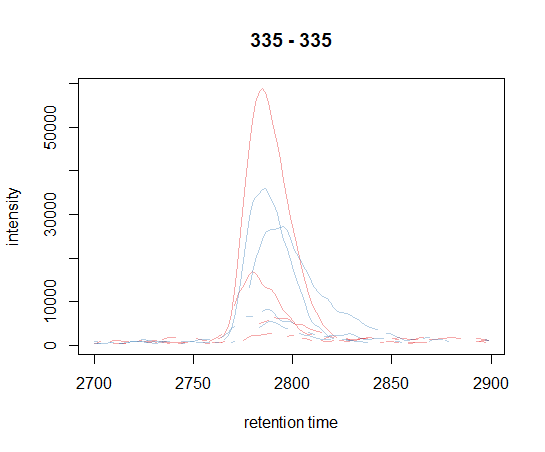
注意,如果在特定扫描(即特定保留时间)中,在各自的mz范围内没有测量到信号,则通过chromatogram法提取的Chromatogram对象包含NA值。反映在上图中为不连续的线。
上面的峰值宽度约为50秒。peakwidth参数应设置为适应数据集中预期的峰值宽度。对于当前示例数据集,我们将其设置为20,80。
对于ppm参数,我们提取对应于上述峰值的完整MS数据(强度、保留时间和m/z值)。为此,我们首先按保留时间过滤原始对象,然后按m/z过滤,最后用type=“XIC”绘制对象以生成下图。我们使用pipe(%>%)命令更好地说明了相应的工作流。还应注意,在这种类型的图中,如果存在色谱峰,则默认显示色谱峰。
1 | raw_data %>% |
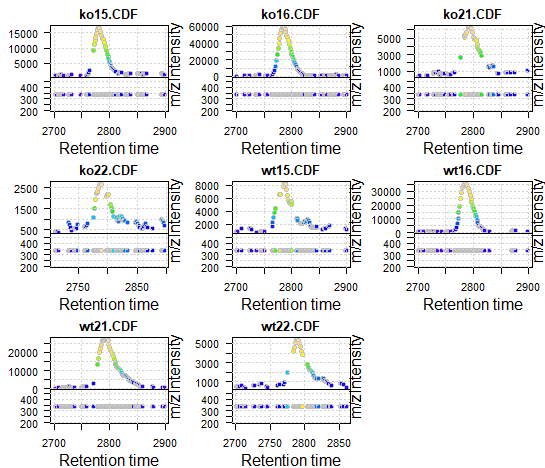
图注:对于每个图:上面板:根据保留时间绘制强度值的色谱图;下面板:m/z根据保留时间绘图。各个数据点根据强度进行着色。
在目前的数据中,m/z值实际上没有变化。通常可以看到m/z值(下面板)散布在化合物的实际m/z值周围。centWave算法的第一步基于连续扫描的m/z值的差异定义所谓的感兴趣区域(regions of interest, ROI)。具体而言,如果ROI的m/z和平均m/z之间的差小于用户定义的ppm参数,则来自连续扫描的m/z值被包括在ROI中。因此,ppm的合理选择可以是来自作为色谱峰一部分的相邻扫描/光谱(scans/spectra)的数据点的最大m/z差。建议检查许多化合物(内标或样品中已知存在的化合物)的m/z值范围,并根据这些确定centWave的ppm参数。
注意,我们也可以对提取的离子色谱图(ion chromatogram)进行峰检测。这有助于评估不同的峰值检测设置。只需注意,提取的离子色谱图上的峰值检测不会考虑ppm参数,并且背景信号的估计与完整数据集上的峰值探测不同;因此,snthresh的值将具有不同的结果。下面我们使用findChromPeaks函数对提取的离子色谱图进行峰值检测。提交的参数对象定义将使用的算法,并允许定义此算法的设置。我们使用带有默认设置的centWave算法,snthresh除外。
1 | xchr <- findChromPeaks(chr_raw, param = CentWaveParam(snthresh = 2)) |
我们可以使用chromPeaks函数访问已识别的色谱峰。
1 | head(chromPeaks(xchr)) |
1 | ## rt rtmin rtmax into intb maxo sn row column |
与chromPeaks矩阵相似,还有一个数据框chromPeakData,允许为每个色谱峰添加任意注释。下面我们提取这个数据框,默认情况下,该数据框只包含识别峰值的MS水平。
1 | chromPeakData(xchr) |
1 | ## DataFrame with 12 rows and 4 columns |
接下来,我们在提取的离子色谱图中绘制已识别的色谱峰。我们使用col参数为各个色谱线上色。还可以为识别的峰值指定颜色,peakCol为前景/边框颜色,peakBg为背景/填充颜色。必须为chromPeaks列出的每个色谱峰提供一种颜色。下面我们定义了一种颜色,以指示样本所在的样本组,并使用峰“sample”列中的样本信息为每个色谱峰分配正确的颜色。更多峰值突出显示选项将在下面进一步描述。
1 | sample_colors <- group_colors[xchr$sample_group] |
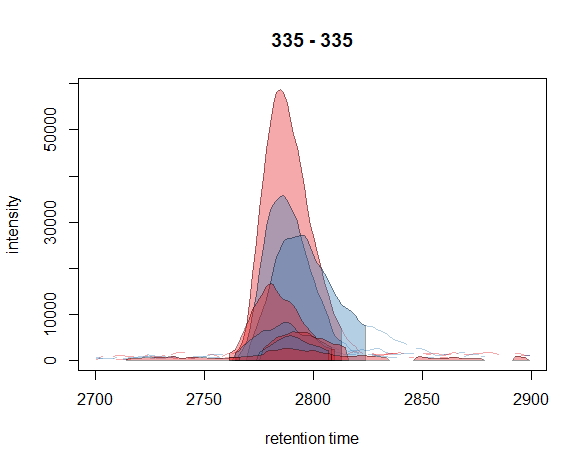
图注:红色和蓝色分别代表KO和野生型样本。已识别色谱峰的峰面积用样品组颜色突出显示。
最后,我们对整个数据集进行色谱峰检测。请注意,我们将参数prefilter设置为c(6, 5000),将noise设置为5000,以减少该示例的运行时间。使用此设置,我们只考虑峰值检测步骤中值大于5000的信号。
1 | cwp <- CentWaveParam(peakwidth = c(20, 80), noise = 5000, |
结果作为XCMSnExp对象返回,该对象通过存储LC/GC-MS预处理结果来扩展OnDiskMSnExp对象。这也意味着,所有设置和过滤数据或访问(原始)数据的方法都是从OnDiskMSnExp对象继承的,因此可以重新使用。还请注意,通过使用参数add=TRUE调用findChromPeaks,可以对xdata对象执行额外的峰值检测(例如,在MS level>1的数据上)。
色谱峰检测的结果可通过chromPeaks方法获得。
1 | head(chromPeaks(xdata)) |
1 | ## mz mzmin mzmax rt rtmin rtmax into intb maxo sn sample |
返回的matrix提供了每个已识别色谱峰的m/z和保留时间范围,以及整合的信号强度(“into”)和最大峰强度(“maxo”)。“sample”列包含确定峰值的对象/实验中样本的索引。
可以使用chromPeakData函数提取每个峰的注释。该数据框还可以用于为每个检测到的峰添加/存储任意注释。
1 | chromPeakData(xdata) |
1 | ## DataFrame with 1707 rows and 2 columns |
峰检测不会总是完美地工作,从而导致峰检测伪影(artifacts),例如重叠峰或人为分割峰。refineChromPeaks函数允许通过去除未通过特定标准的已识别峰或合并人工拆分的色谱峰来优化峰检测结果。使用参数对象CleanPeaksParam和FilterIntensityParam,可以分别删除保留时间范围或强度低于阈值的峰(有关更多详细信息和示例,请参阅各自的帮助页面)。使用MergeNeighbringPeaksParam可以合并色谱峰。下面我们对峰检测结果进行进一步处理,如果它们之间的信号低于较小峰最大强度的75%,合并每个文件在4秒窗口中重叠的峰值(merging peaks overlapping in a 4 second window per file)。有关设置和方法的详细说明,请参阅MergeNeighborgingPeaksParam帮助页面。
1 | mpp <- MergeNeighboringPeaksParam(expandRt = 4) |
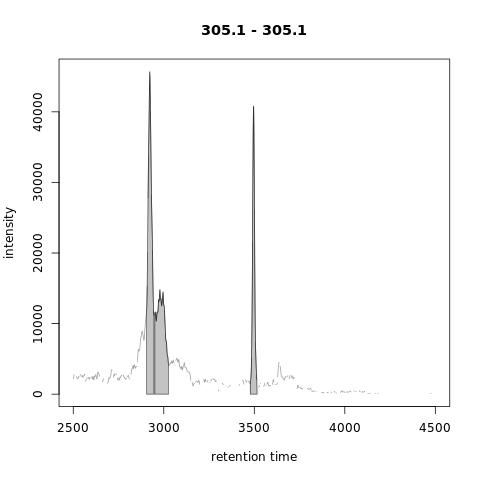
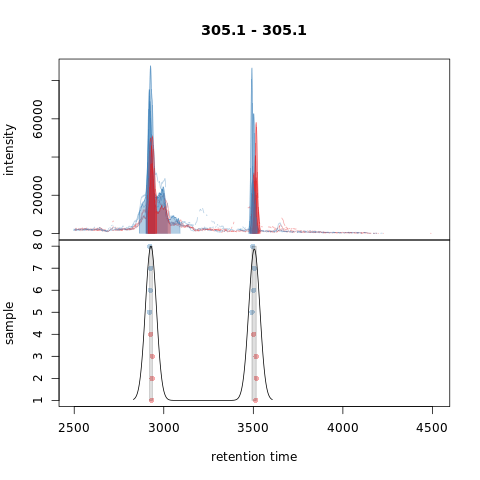
如下展示了合并峰的示例。
1 | mzr_1 <- 305.1 + c(-0.01, 0.01) |
 |
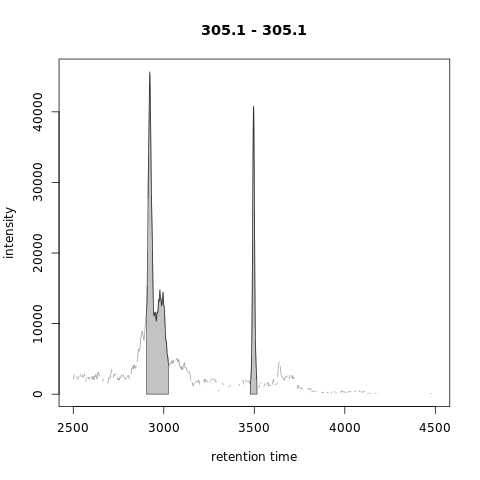 |
对于以上色谱图centWave检测到3个峰(1个峰代表整个区域,两个较小的峰,见上图中的左侧面板)。MergeNeighborgingPeaksParam的峰细化(refinement )将它们减少为单个峰(上图中的右面板)。注意,这种细化不会合并相邻峰值,因为它们之间的信号低于一定比例(参见下图)。
1 | mzr_1 <- 496.2 + c(-0.01, 0.01) |
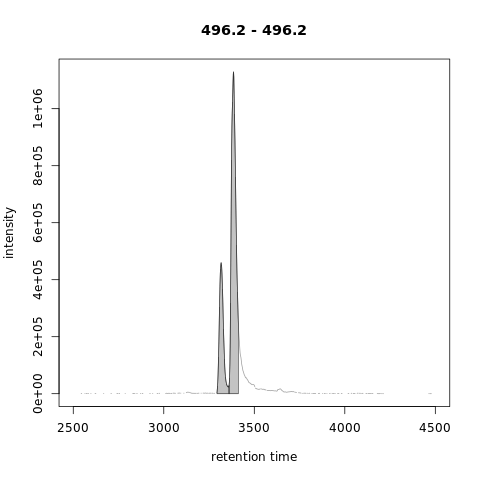 |
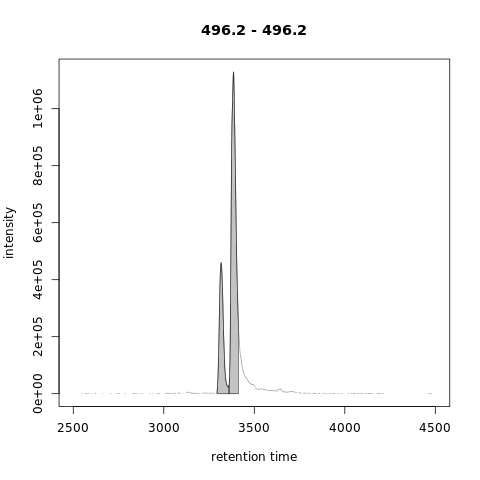 |
还应注意,可以对提取的离子色谱图进行峰细化。这可以例如用于微调参数的设置。为了说明这一点,我们下面对提取的离子色谱图chr_1进行了峰细化,减少了minProp参数,以强制连接两个峰。
1 | res <- refineChromPeaks(chr_1, MergeNeighboringPeaksParam(minProp = 0.05)) |
1 | ## mz mzmin mzmax rt rtmin rtmax into intb maxo sn |
1 | png(file="cxms_11.png") |
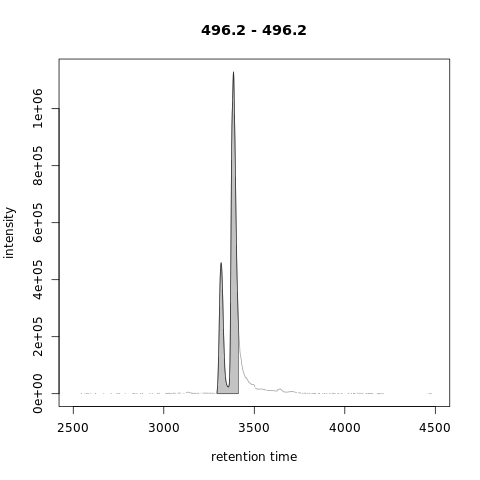
在继续之前,我们用峰细化的结果替换xdata对象。
1 | xdata <- xdata_pp |
下面我们使用chromPeaks矩阵中的数据来计算每个文件的一些摘要。
1 | summary_fun <- function(z) |
1 | ------------------------------------------------------------------------ |
我们还可以使用plotChromPeaks函数绘制一个文件的m/z——保留时间空间中已识别色谱峰的位置。下面我们绘制了第三个样品的色谱峰。
1 | png(file="xcms_12.png") |
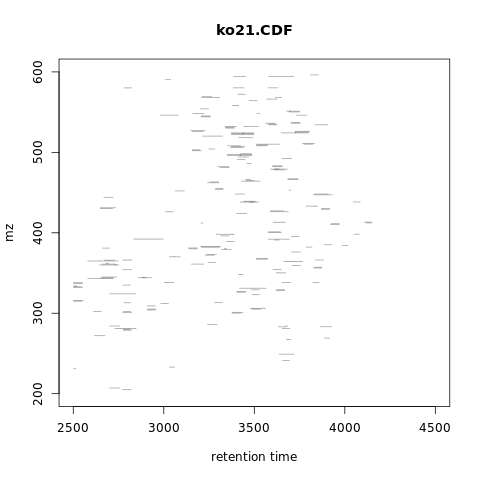
为了获得峰检测的全局概览,我们可以沿着保留时间轴绘制每个文件的已识别峰的频率。这允许识别MS运行中的时间段,在该时间段中识别出更多数量的峰,并评估这在文件之间是否一致。
1 | png(file="xcms_13.png") |
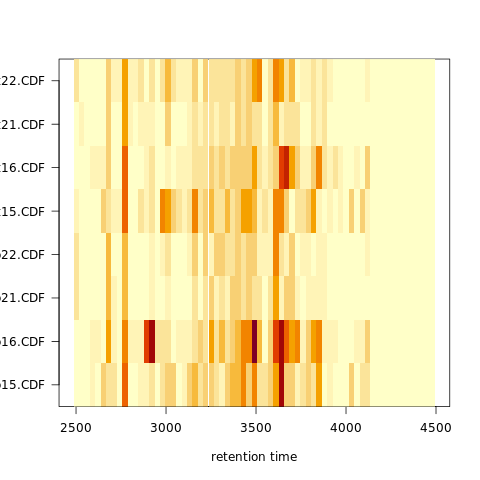
图注:频率是彩色编码的,较高的频率由黄-白表示。每行显示一个文件的峰值频率。
接下来,我们重点介绍前面示例峰的已识别色谱峰。在对应于已知峰或内标的峰列表上评估这样的图有助于确保峰检测设置是适当的并且正确地识别预期峰。我们从峰检测结果对象中提取离子色谱图,其中还包含我们可以使用chromPeaks函数提取的该离子的已识别色谱峰。
1 | chr_ex <- chromatogram(xdata, mz = mzr, rt = rtr) |
1 | ## mz mzmin mzmax rt rtmin rtmax into intb maxo sn |
我们还可以绘制提取的离子色谱图。已识别的色谱峰将在图中自动突出显示。下面我们用矩形突出显示色谱峰,从峰的最小到最大rt,从0到峰的最大信号。
1 | sample_colors <- group_colors[chr_ex$sample_group] |
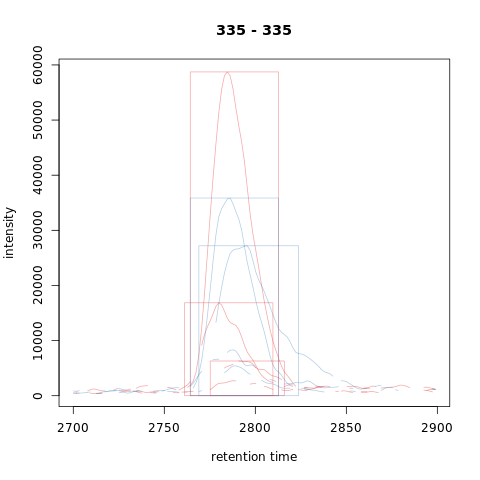
图注:红色和蓝色分别代表KO和野生型样本。矩形表示每个样品的色谱峰。
作为上述矩形可视化的替代方案,可以用一个点表示每个峰的顶点位置(将参数type="point"传递给函数),或通过指定type="polygon"绘制实际识别的峰。为了完全省略突出显示已识别的峰(例如绘制基本峰色谱图或类似图),可以使用type = "none"”。下面我们使用type="polygon"填充每个样品中每个已识别色谱峰的峰面积。然而,在这样的图中是否仍然可以识别单个峰取决于从中绘制峰的样本数量。
1 | png(file="xcms_15.png") |
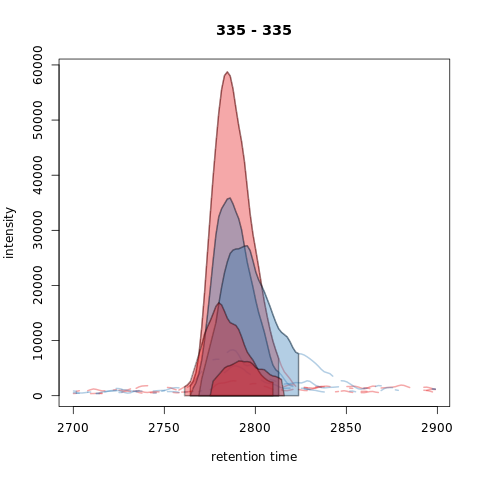
图注:红色和蓝色分别代表KO和野生型样本。已识别色谱峰的信号区域用颜色填充。
注意,我们还可以通过在chromPeaks方法中提供各自的m/z和保留时间范围以及mz和rt参数,提取选定区域的已鉴定色谱峰。
1 | pander(chromPeaks(xdata, mz = mzr, rt = rtr), caption = paste("Identified chromatographic peaks in a selected ", "m/z and retention time range.")) |
1 | ----------------------------------------------------------------------------- |
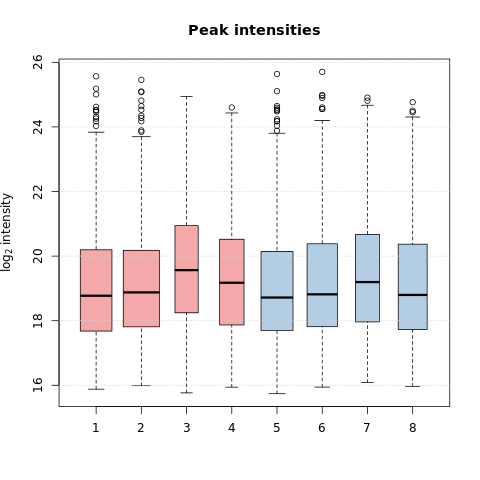
最后,我们还绘制了每个样本的峰强度分布。这允许调查样本之间的峰信号是否存在系统差异。
1 | ## Extract a list of per-sample peak intensities (in log2 scale) |
注意,除了上述色谱峰的识别之外,还可以使用manualChromPeaks函数手动定义和添加色谱峰(更多信息请参见?manualChronPeaks帮助页面)。
Alignment
分析物在色谱中洗脱的时间可能因样品(甚至化合物)而异。在上一节中作为示例所示的提取离子色谱图中已经可以观察到这种差异。对齐步骤,也称为保留时间校正,旨在通过沿保留时间轴移动信号来调整这一点,以在实验中对齐不同样本之间的信号。
存在很多的对齐算法(参见[3]),其中一些算法也在xcms中实现。在xcms中执行对齐/保留时间校正的方法是adjustRtime,其根据所提供的参数类别使用不同的对齐算法。
在下面的示例中,我们使用obiwarp方法[4]来对齐样本。我们使用binSize=0.6,它在mz为0.6的bins中创建扭曲函数(warping functions)。此外,这里建议修改每个实验的设置,并评估保留时间校正是否对齐了内参或已知化合物。
1 | xdata <- adjustRtime(xdata, param = ObiwarpParam(binSize = 0.6)) |
adjustRtime,除了计算每个光谱的校正保留时间外,还调整了已鉴定色谱峰的报告保留时间。
为了提取校正后的保留时间,我们可以使用adjustedRtime方法,或者简单地使用rtime方法,如果存在的话,默认情况下会从XCMSnExp对象返回校正后的保留时间。
1 | ## Extract adjusted retention times |
1 | ## F1.S0001 F1.S0002 F1.S0003 F1.S0004 F1.S0005 F1.S0006 |
1 | ## Or simply use the rtime method |
1 | ## F1.S0001 F1.S0002 F1.S0003 F1.S0004 F1.S0005 F1.S0006 |
原始保留时间可以从包含与rtime(xdata,adjusted=FALSE)对齐的数据的XCMSnExp中提取。
为了评估对齐的影响,我们利用校正后的数据绘制BPC。此外,我们使用plotAdjustedRtime函数绘制每个样本的校正后保留时间与原始保留时间的差异。对于基峰色谱图,从结果对象中提取已识别的色谱峰是没有意义的。因此,我们在chromatogram调用中使用参数include="none",以在返回的对象中不包含色谱峰。请注意,也可以通过在plot调用中设置peakType = "none"来避免绘制它们。
1 | ## Get the base peak chromatograms. |
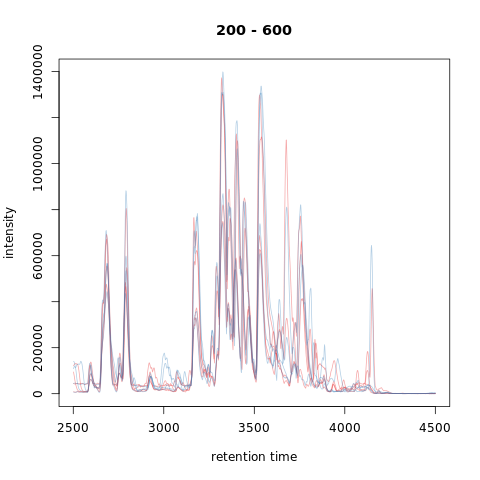
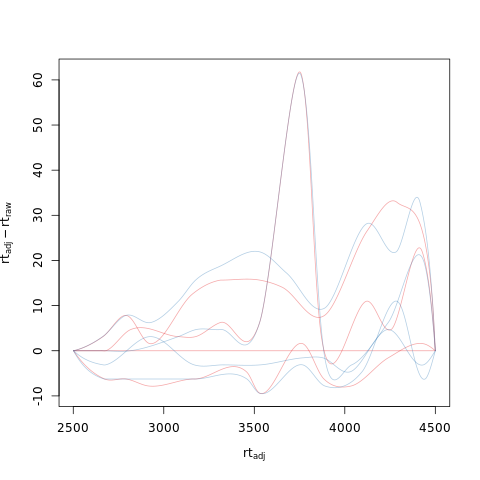
图注:对齐后的基峰色谱图(顶部)和沿保留时间轴的校正保留时间与原始保留时间之间的差异(底部)。
校正后的保留时间与原始保留时间之间的差异太大,可能表明样品或对齐效果不佳(poorly performing samples or alignment)。
注意:XCMSnExp对象保存原始数据以及校正后的保留时间,在大多数情况下,子设置(subsetting)将删除校正后的保持时间。因此,一些情况下用校正后的保留时间代替原始保留时间可能是有用的。这可以通过applyAdjustedRtime完成。
最后,我们评估了对齐对测试峰的影响。
1 | par(mfrow = c(2, 1)) |
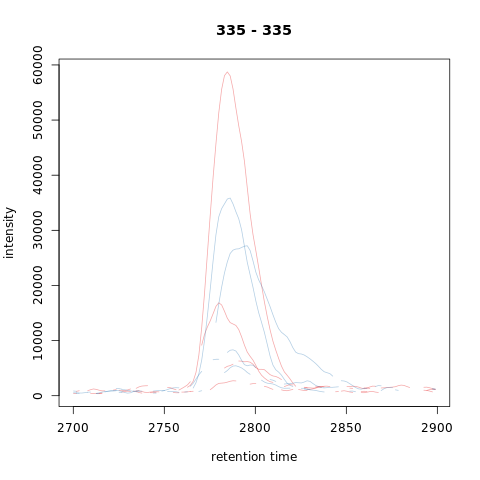
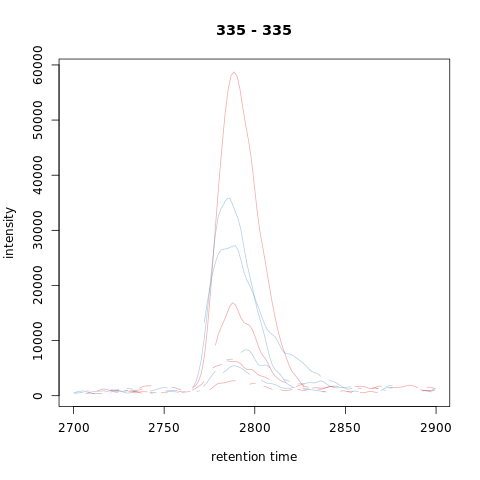
Subset-based alignment
在一些实验中,仅根据样本的子集进行比对可能会有所帮助,例如,如果QC样本定期注射(injected ),或者如果实验包含空白样本。xcms中的对齐方法允许估计样本子集(不包括空白样本的所有样本或在测量运行期间定期注入的质控样本)的保留时间漂移,并使用这些来调整整个数据集。
参数subset(PeakGroupsParam或ObiwarpParam对象)可用于定义样本子集,整个数据集的对齐将基于该样本子集(例如,子集是QC样本的索引),参数subsetAdjust允许指定调整遗漏样本(left-out samples)的方法。目前有两个选项可用:
subsetAdjust = "previous":根据先前子集样本(例如QC样本)的对齐结果调整非子集样本的保留时间。如果样本的顺序为A1、B1、B2、A2、B3、B4,A代表QC样本,B代表研究样本,则使用subset = c(1, 4)和subsetAdjust = "previous"将导致所有A样本彼此对齐,而非子集样本B1和B2将根据子集样本A1和B3以及B4对A2样本的对齐结果进行调整。subsetAdjust = "average":基于先前和后续子集样本的对齐结果的内插(interpolation)来调整非子集样本的保留时间。在上述示例中,B1将基于子集(QC)样本A1和A2之间的校正保留时间的平均值进行调整。由于在非子集样本B3和B4之后没有子集样本,因此将仅基于A2的对齐结果来校正这些样本。注意,加权平均值用于计算经校正的保留时间平均值,其使用非子集样本与子集样本的索引之差的倒数作为权重。因此,如果我们有类似A1、B1、B2、A2的设置,在校正非子集样本B1时,A1的校正保留时间将比A2的更大,从而使其校正保留时间更接近A1的保留时间,而不是A2的保留时间。请参见下面的示例。
这两种情况都需要对对象内的样本进行有意义的/正确的排序(例如按注入索引排序)。
以下示例旨在说明这些对齐选项的效果。我们假设faahKO数据集中的样本1、4和7是质控样本(样本池)。因此,我们希望基于这些样本执行对齐,随后基于来自相邻子集(QC)样本的结果的插值来校正剩余的样本(2、3、5、6和8)的保留时间。在初始峰分组后,我们使用峰组法(peak groups method)进行校准,通过子集参数传递我们希望校准所基于的样本的索引,并指定subsetAdjust = "average"以根据相邻子集/QC样本的校准结果的插值校正研究样本。
注意,对于任何子集对齐,所有参数(如minFraction)都是相对于子集的,而不是整个实验!
要重新执行对齐,我们可以首先使用dropAdjustedRtime函数删除以前的对齐结果。
1 | xdata <- dropAdjustedRtime(xdata) |
接下来,我们必须进行初始对应分析,因为峰群对齐方法通过对齐之前识别的hook峰(存在于大多数/所有样品中的色谱峰;下一节将介绍所用算法的详细信息)来校正保留时间。我们在这里使用默认设置,但强烈建议为每个数据集调整参数。PeakDensityParam必须定义样本组(即,将单个样本分配给实验中的样本组)。如果实验中没有样本组,则应将每个文件的样本组设置为单个值(例如rep(1, length(fileNames(xdata)))。
1 | ## Initial peak grouping. Use sample_type as grouping variable |
下面,我们绘制了对齐结果,用绿色标记了作为子集一部分的样本,其他样本用灰色标注。这很好地显示了subsetAdjust = "average"的插值是如何工作的:样本2的保留时间是根据子集样本1和4的保留时间进行校正的,但是,给更接近的子集样本1赋予了更大的权重,这导致校正后的样本2的保持时间与样本1的保持时间更相似。另一方面,样本3得到校正,给第二子集样本(4)赋予更多权重。
1 | clrs <- rep("#00000040", 8) |
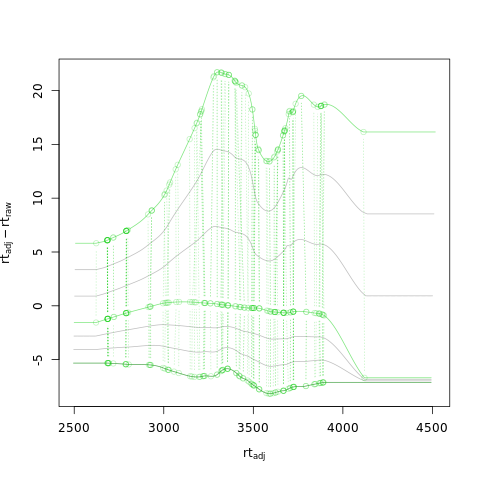
图注:沿保留时间轴校正保留时间与原始保留时间之间的差异。对校准模型进行估算的样本显示为绿色,研究样本显示为灰色。
选项subsetAdjust = "previous"基于单个子集样本(先前的)校正非子集样本的保留时间,这在大多数情况下导致非子集样本校正后的保留时间与用于校正的子集样本高度相似。
Correspondence
代谢组学预处理的最后一步是匹配样本之间(取决于设置,如果样本相邻,也在样本内)检测到的色谱峰的对应关系。在xcms中执行对应的方法是groupChromPeaks。我们将使用峰密度法[5]对色谱峰进行分组。该算法结合了色谱峰,这取决于沿着mz维度的小切片中沿着保留时间轴的峰密度。为了说明这一点,我们绘制了每个样品中具有多个色谱峰的mz切片的色谱图。我们使用0.4的minFraction参数值,因此每个样本组中至少有40%的样本中存在的色谱峰被分组为特征(feature)。样品组分配是使用sampleGroups参数指定的。
1 | ## Define the mz slice. |
图注:上图:具有多个色谱峰的mz切片的色谱图。下图:确定保留时间的色谱峰(x轴)和不同bw参数值的实验样品内的指数(y轴)。黑线表示峰密度估计值。峰分组(基于提供的设置)由灰色矩形表示。
上图中的上部面板显示了每个样品的提取离子色谱图,突出显示了检测到的峰。中间和下部曲线显示了不同样品中每个检测到的峰的保留时间。黑色实线表示检测到的峰沿保留时间的密度分布。组合成特征(峰值组)的峰值用灰色矩形表示。这种类型的可视化非常适合在将示例m/z切片应用于完整数据集之前测试它们的对应设置。
下面,我们将基于定义设置(defined settings)对整个数据集进行对应分析。
1 | ## Perform the correspondence |
基于xcms的预处理结果可以通过quantify法从SummarizedExperiment包中汇总为SummarizedExperiment对象。该对象将包含作为分析矩阵的特征丰度、作为rowData(即行注释)的特征定义(其m/z、保留时间和其他元数据)和作为colData(即列注释)的样本/表型信息。所有处理历史记录都将放入对象的元数据中(metadata)。然后,该对象可以用于任何进一步的(与xcms无关的)处理和分析。
下面我们使用quantify来生成当前分析的结果对象。参数值和任何其他附加参数将传递给featureValues方法,该方法在内部用于创建特征丰度矩阵。
1 | res <- quantify(xdata, value = "into") |
样品注释可以通过colData方法访问。
1 | colData(res) |
1 | ## DataFrame with 8 rows and 3 columns |
通过rowData注释特征:
1 | rowData(res) |
1 | DataFrame with 357 rows and 11 columns |
特征丰度可以通过assay方法获得。还要注意,SummarizedExperiment支持多个这样的分析矩阵。
1 | head(assay(res)) |
1 | ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF |
此外,还可以使用featureDefinitions和featureValues方法分别从对应分析中提取结果,前者返回带有特征定义的数据框(即mz和rt范围,在列peakidx中为每个特征的chromPeaks矩阵中的色谱峰指数),后者为特征丰度。
1 | ## Extract the feature definitions |
1 | DataFrame with 357 rows and 11 columns |
featureValues方法返回一个矩阵,其中行是特征,列是样本。这个矩阵的内容可以使用value参数定义。默认值value = "into"返回一个矩阵,其中包含样本中某个特征对应的峰的整合信号。chromPeaks矩阵的任何列名都可以传递给参数value。下面我们提取每个特征/样本的整合峰值强度。
1 | ## Extract the into column for each feature. |
1 | ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF |
该特征矩阵包含某些样品在特征m/z-rt区域未检测到色谱峰(NA值)。虽然在许多情况下,在相应的区域可能确实没有峰信号,但也可能有信号,但峰检测算法未能检测到色谱峰(例如,因为信号太低或噪声太大)。xcms提供fillChromPeaks方法来填充原始文件中这些缺失值的强度数据。填充的峰被添加到chromPeaks矩阵中,并在chromPeakData数据框的“is_filled”列中用TRUE表示。下面我们执行这样的填充。
1 | xdata <- fillChromPeaks(xdata, param = ChromPeakAreaParam()) |
1 |
|
对于样本中没有检测到峰的特征,该方法提取特征的mz-rt区域中的所有强度,对信号进行整合(integrates),并将填充的峰添加到chromPeaks矩阵中。如果特征的mz-rt区域没有测量/可用信号,则不添加峰。对于这些,即使在填写了缺失的峰数据后,也会在featureValues矩阵中报告NA。
可以使用不同的选项来定义特征的mz-rt区域。使用上面使用的ChromPeakAreaParam()参数对象,使用其所有(检测到的)色谱峰的m/z和rt范围定义特征区域:区域中低的m/z值定义为特征所有峰的“mzmin”值的下四分位(25%分位数),高m/z值为“mzmax”值的上四分位数(75%分位数),低rt值作为“rtmin”值的下四分位(25%分位数),高rt值为“rtmax”值的上四分位数(75%分位数)。这确保了信号是从特定特征区域集成的。
或者,可以在fillChromPeaks调用中使用FillChromPpeaksParam参数对象,这类似于初始(old)xcms实现的方法。
下面我们比较填写缺失值前后缺失值的数量。我们可以使用featureValues方法的filled参数来定义是否也应该返回填充的峰值(filled-in peak values)。
1 | ## Missing values before filling in peaks |
1 | ## ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF wt22.CDF |
1 | ## Missing values after filling in peaks |
1 | ## ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF wt22.CDF |
接下来,我们使用featureSummary函数获取每个特征的一般摘要,其中包括找到峰的样本数量或为特征分配了多个峰的样本数。同时指定样本组可以分解每个样本组的汇总统计信息。
1 | head(featureSummary(xdata, group = xdata$sample_group)) |
1 | count perc multi_count multi_perc rsd KO_count KO_perc |
我们可以将特征值矩阵与缺失峰值的填充数据一起添加到我们的SummarizedExperiment对象res中,作为附加矩阵(assay):
1 | assays(res)$raw_filled <- featureValues(xdata, filled = TRUE) |
我们现在有两个可用的矩阵(assays),一个具有检测值的矩阵,一个包含检测值和填充值的矩阵,每个都可以通过其名称访问。
1 | assayNames(res) |
1 | ## [1] "raw" "raw_filled" |
1 | head(assay(res, "raw")) |
1 | ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF |
1 | head(assay(res, "raw_filled")) |
1 | ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF |
应始终通过检查提取的离子色谱图(例如已知化合物、内标或一般识别特征)来评估峰检测、比对(alignment)和对应(correspondence)的性能。featureChromatogram函数允许提取featureDefinitions中每个特征的色谱图。返回的MCchromatograms对象包含每个特征(每行包含一个特征的数据)和样本(每列代表一个样本的数据)的离子色谱图。下面我们提取前4个特征的色谱图。
1 | feature_chroms <- featureChromatograms(xdata, features = 1:4) |
1 | ## XChromatograms with 4 rows and 8 columns |
绘制提取的离子色谱图。我们再次使用每个鉴定峰的组颜色来填充该区域。
1 | png(file="xcms_24.png") |
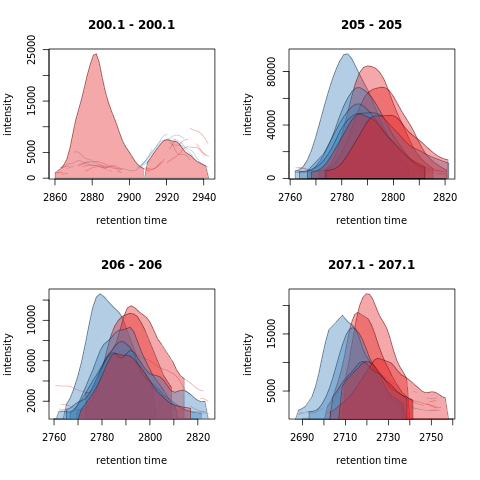
为了访问第二个特征的EIC,我们可以简单地将feature_chros对象取子集。
1 | eic_2 <- feature_chroms[2, ] |
1 | mz mzmin mzmax rt rtmin rtmax into intb maxo sn |
最后,我们进行了主成分分析,以评估本实验中样本的分组。注意,我们没有执行任何数据标准化,因此分组可能(也将)受到技术偏见的影响。
1 | ## Extract the features and log2 transform them |
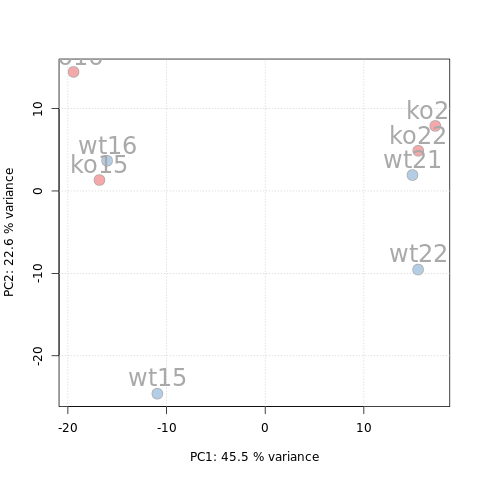
我们可以在PC2上看到KO和WT样品之间的预期分离。在PC1上样本根据其ID进行分离,ID<=18的样本与ID>18的样本被分开。这种分离可能是由于技术偏差(例如,在不同的天/周进行的测量)或由于所分析小鼠的生物学特性(性别、年龄、产仔等)造成的。
Further data processing and analysis
标准化特征的信号强度是必需的,但目前xcms还不支持(在不久的将来可能会添加一些方法)。建议将quantify方法返回的SummarizedExperiment用于任何进一步的数据处理,因为这种类型的对象在同一对象中存储特征定义(feature definitions)、样本注释以及特征丰度。为了识别例如具有显著不同强度/丰度的特征,建议使用其他R包中提供的功能,例如Bioconductor的limma包。为了支持依赖于旧xcmsSet结果对象的其他包,可以使用xset <- as(x, "xcmsSet")将新的XCMSnExp对象强制转换为xcmsSet对象,其中x是XCMSnExp对象。
参考
关注公众号“生信之巅”。
 |
 |
敬告:使用文中脚本请引用本文网址,转载请注明出处请尊重本人的劳动成果,谢谢!



