
2025年9月,InstaDeep(英国伦敦)、BioNTech(德国美因茨)、哥本哈根大学计算机系等机构的Bernardo P. de Almeida、Hugo Dalla-Torre(共同第一作者)、Thomas Pierrot(通讯作者)等研究者在《Nature Methods》期刊发表了题为“Annotating the genome at single-nucleotide resolution with DNA foundation models”的学术文章。该文章报道了其开发的基因组注释模型家族(SegmentNT、SegmentEnformer、SegmentBorzoi),在基因组功能元件注释领域具有突破传统工具局限性、实现14种基因及调控元件单核苷酸分辨率精准注释的重要意义。
1 前言
1.1 研究背景
基因组注释模型是现代生物学研究的核心工具,可直接从DNA序列中识别基因、外显子-内含子结构及其他功能元件。随着测序技术发展,已测序基因组数量呈指数增长,精准高效的DNA序列注释不仅有助于解析遗传结构,还对遗传变异预测、计算机模拟序列设计等应用至关重要。
现有注释工具存在明显局限:基于隐马尔可夫模型的工具(如AUGUSTUS)虽具备单核苷酸分辨率,但难以建模生物复杂性,预测基因异构体和全染色体注释时需依赖实验数据;调控元件识别工具多针对特定元件单独开发,训练数据集规模有限且分布与实际应用场景差异大,泛化能力不足。
DNA基础模型(参数达数亿至数十亿,训练数据达数千亿至数万亿token)的出现为解决上述问题提供了新思路,其可通过无监督或监督训练学习通用序列表征,适配多种下游任务。
1.2 研究目标
将基因组注释问题构建为多标签语义分割任务,利用预训练DNA基础模型微调,开发能在单核苷酸分辨率下注释14种基因及调控元件的通用模型,突破传统工具的元件特异性和序列长度限制,提升跨物种泛化能力。
2 研究方法
2.1 基因组分割模型架构
2.1.1 SegmentNT架构
SegmentNT以预训练DNA基础模型Nucleotide Transformer(NT-Multispecies-v2,500M参数)为DNA编码器,采用6-mer分词器提取序列嵌入特征(序列长度N与token数L满足$L≈N/6$)。模型替换NT的原始语言模型头,引入1D U-Net分割头,包含2个下采样卷积块和2个上采样卷积块,各块分别含2048和4096个卷积核,序列长度分别为 $L/2$ 和 $L/4$,总参数6300万。
U-Net输出张量形状为$(N, K, 2)$(K=14种元件),经softmax层后得到每个核苷酸属于每种元件的概率$P$及非概率$1-P$。模型允许单个核苷酸属于多种元件,二元分类阈值设为0.5。
2.1.2 SegmentEnformer与SegmentBorzoi架构
将Enformer和Borzoi作为DNA编码器,其原始架构含卷积-下采样块及自注意力块(Enformer分辨率128 bp,Borzoi分辨率32 bp)。在两模型最后一层表征(预测头前)添加U-Net分割头,分别命名为SegmentEnformer和SegmentBorzoi,保持与SegmentNT一致的训练和验证超参数。
2.2 模型训练与评估
2.2.1 训练参数
- 优化器:Adam,学习率$5×10^{-5}$
- 批次大小:256
- 训练数据量:SegmentNT-3kb模型训练102.4亿token(2048万条序列),10kb、20kb、30kb模型基于前一尺寸最佳检查点初始化并微调(如30kb模型额外训练25.6亿token,51万条序列)
- 损失函数:焦点损失($γ=2$),聚焦稀疏元件对应的“困难样本”
- 硬件:8块H100 GPU,训练时长20小时(3kb模型)
2.2.2 数据集分割
按染色体划分训练集、验证集和测试集:20、21号染色体为测试集,22号为验证集,其余为训练集。排除测试集中与训练/验证集基因同源的片段(未剔除可能影响这些区域性能的同源远端调控元件),验证集和测试集采用固定滑动窗口采样,测试集进行10次随机采样以计算置信区间。
2.2.3 评估指标
- 核苷酸水平指标:马修斯相关系数(MCC)、精确率-召回率曲线下面积(auPRC)、雅卡尔相似度(Jaccard)、F1分数
- 区域水平指标:片段重叠分数(SOV),使用默认λ=1.0 (代码: http://dna.cs.miami.edu/SOV/)
2.3 模型消融与基线设置
2.3.1 消融实验模型
- 以NT v1 2.5B 1000G模型为骨干(总参数26亿)
- 仅微调U-Net分割头的SegmentNT-3kb模型(5.63亿参数)
- 编码器随机初始化的SegmentNT模型(全参数训练或仅训练分割头)
- 直接输入one-hot编码的U-Net模型(6300万或2.52亿参数)
- one-hot编码经线性层扩展至1024维嵌入的U-Net模型(6600万参数)
2.3.2 基线模型
- BPNet:2个版本(嵌入维度64对应12万参数,1024对应2900万参数)
- SpliceAI:3个版本(嵌入维度32对应70万参数,256对应4400万参数,920对应5.73亿参数)
2.4 上下文长度扩展方法
由于SegmentNT的DNA编码器采用的旋转位置编码(RoPE)在训练时的最大序列长度为2048个token,因此在对更长序列进行推理时,其性能会迅速下降。此前已有多项研究提出了对RoPE的改进方案,以更好地处理长序列的评估或微调任务,例如采用位置插值法或“NTK感知的缩放旋转位置编码”(NTK-aware scaled RoPE)。
Peng等人提出了一种适用于未见过序列长度的RoPE适配方案,名为YaRN。经过测试,与直接使用“NTK感知的缩放旋转位置编码”相比,YaRN在扩展SegmentNT的序列长度方面并未带来性能提升。由于后者的实现更为简便,作者最终选择采用该方法来扩展SegmentNT的上下文长度。
设隐藏层神经元集合为(D),序列向量为$(x_{1}, …, x_{L} \in \mathbb{R}^{|D|})$,则“NTK感知的旋转位置编码”可通过以下公式描述:
$f^′_{w}(x_{m}, m, θ_{d}) = f_{w}(x_{m}, g(m), h(θ_{d}))$
其中,(d)表示嵌入维度上的位置,(m)表示嵌入在序列中的位置,(J)为RoPE函数,(w)表示可学习参数(权重),$(g(m)=m)$,$(h(\theta_{d})=b^{\prime-\frac{2d}{|D|}})$,$(b’=b \cdot s^{\frac{|D|}{|D|-2}})$,最终满足$(\frac{2\pi}{\theta_{d}}=2\pi b^{\frac{2d}{10}})$。
为完整说明,(b)是旋转位置编码中使用的指数基,(b’)是对(b)进行缩放后的版本,用于适配NTK感知的缩放逻辑和上下文长度。缩放因子(s)的计算方式为$(s=\frac{L’}{L})$,其中(L’)为扩展后的上下文长度,(L)为训练时的上下文长度(对于NT-Multispecies-v2(500M)模型,(L=2048)个token)。
对于采用“NTK感知的旋转位置编码”训练的SegmentNT模型,所有长度小于其训练长度的序列,在评估时均使用训练过程中采用的同一缩放因子。具体而言,SegmentNT-30kb模型的训练缩放因子(s=2.44),因此对长度小于30,000 bp的序列进行推理时,仍使用(s=2.44);而对50 kb序列进行评估时,缩放因子则调整为(s=4.07)。
2.5 多物种训练
基于人类SegmentNT-30kb模型,加入小鼠(mm10)、鸡(galGal6)、果蝇(dm6)、斑马鱼(danRer11)、秀丽隐杆线虫(ce11)的注释数据进行微调,得到多物种模型。各物种数据权重:人类5、小鼠4、鸡/果蝇/斑马鱼2、线虫1。各物种单独划分验证集和测试集(如小鼠:验证集chr19,测试集chr18)。
2.6 基因组注释数据来源
2.6.1 人类基因组元件数据
14种元件分为基因元件(蛋白质编码基因、长链非编码RNA、5’UTR、3’UTR、外显子、内含子、剪接受体位点、剪接供体位点)和调控元件(多聚腺苷酸信号、组织不变型/组织特异性启动子、组织不变型/组织特异性增强子、CTCF结合位点)。基因元件和多聚腺苷酸信号来自GENCODE V44注释(排除三级转录本),调控元件来自ENCODE的SCREEN数据库。
2.6.2 多物种数据集
聚焦7种核心基因元件(蛋白质编码基因、5’UTR、3’UTR、外显子、内含子、剪接受体位点、剪接供体位点),注释数据来自Ensembl数据库。测试集含10种动物(如野牛、鲸鱼、猫等)和5种植物(拟南芥、大豆、水稻、小麦、玉米)。
2.7 基准测试方法
2.7.1 基因注释基准
与AUGUSTUS在三种场景对比:30kb基因片段(仅主异构体)、30kb基因片段(所有异构体)、全染色体(所有异构体),评估指标为F1分数、MCC、精确率、召回率、SOV。
2.7.2 剪接位点预测基准
与SpliceAI、Pangolin在两种测试集对比:SpliceAI的mRNA测试集(适配30kb窗口,移除含N序列)、SegmentNT的全染色体测试集(仅保留正义链基因),评估指标为auPRC、MCC、top-k准确率。
2.7.3 调控元件定位基准
与滑动窗口方法(NT微调模型、DeePromoter)对比,将组织不变型和组织特异性启动子/增强子合并为单一类别,评估指标为auPRC,计算单A100 GPU上的推理时间。
3 实验结果
3.1 SegmentNT:DNA序列核苷酸分辨率分割模型
3.1.1 模型性能基础表现
SegmentNT-3kb在14种元件上表现出高核苷酸分辨率定位能力,外显子、剪接位点、3’UTR和组织不变型启动子的测试MCC均高于0.5,长链非编码RNA和CTCF结合位点最难预测(MCC低于0.1)。
SegmentNT-10kb(平均MCC 0.42)性能优于3kb版本(0.37),蛋白质编码基因、3’UTR、外显子和内含子的提升尤为显著,表明这些元件依赖更长序列上下文(如图1c所示)。
3.1.2 基因位点注释示例
在包含NOP56基因(正义链)和IDH3B基因(反义链)的10kb窗口中,SegmentNT-10kb准确预测了两种基因的蛋白质编码属性、5’UTR和3’UTR位置、剪接位点、外显子-内含子结构及多聚腺苷酸信号,同时捕获了NOP56基因的组织特异性和组织不变型启动子,以及区域内的多个增强子(如图1d所示)。
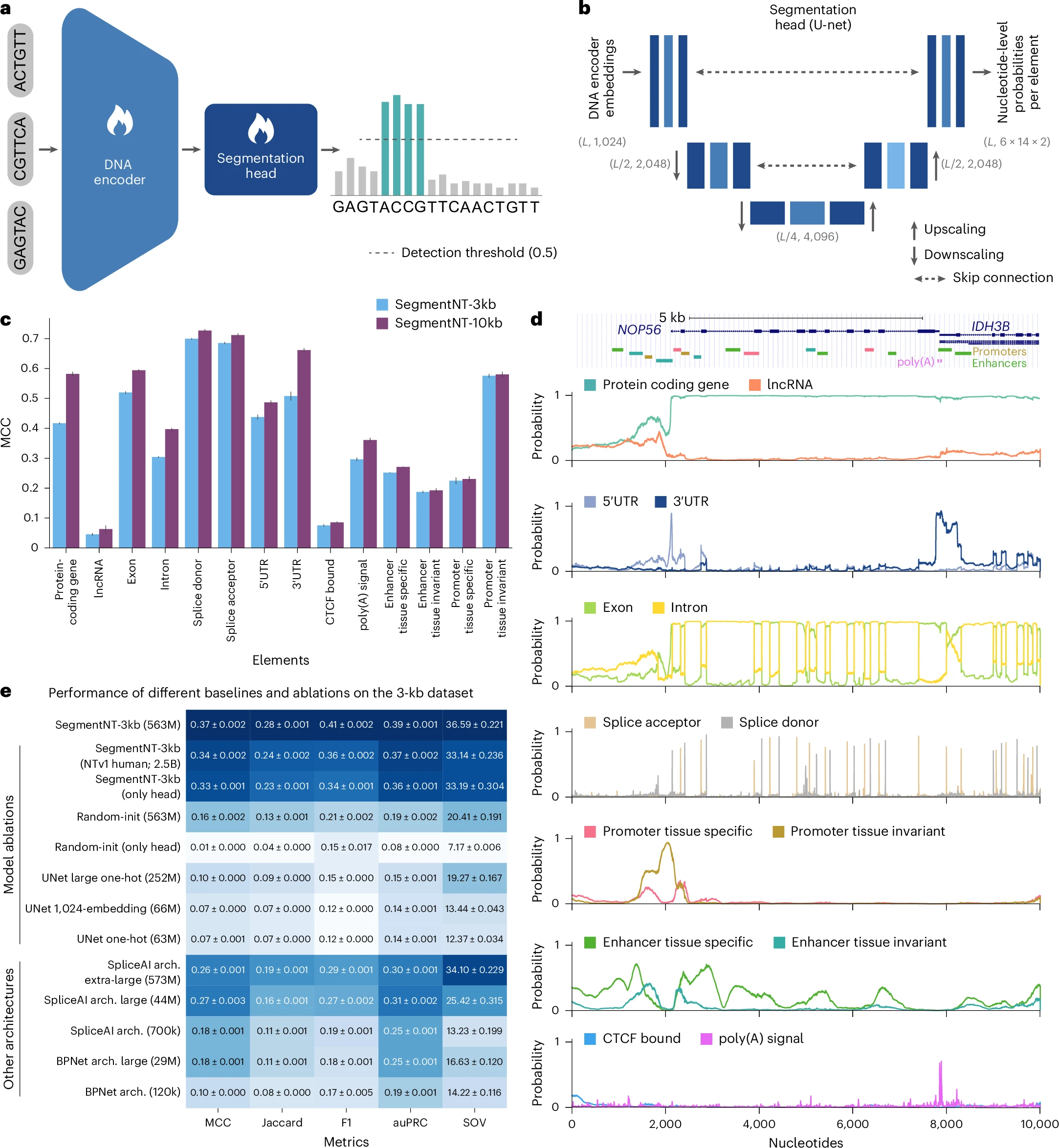
图注:a. SegmentNT神经网络架构,含预训练DNA编码器(NT22)和U-Net分割头,输出各基因组元件的核苷酸分辨率概率;b. 1D U-Net分割头结构,含2个下采样和2个上采样卷积块及跳跃连接,标注各层维度(N为核苷酸数,L为DNA token数,$L≈N/6$);c. SegmentNT-3kb和10kb模型在14种基因组元件上的MCC性能(数据为10次测试集采样的均值±95%置信区间);d. NOP56/IDH3B基因位点的14种元件注释及预测概率示例,含基因异构体、外显子-内含子结构及调控元件;e. SegmentNT与不同消融模型和架构的性能对比(指标为MCC、Jaccard、F1、auPRC、SOV,数据为14种元件的均值±标准差,列归一化颜色标度)。
3.2 预训练DNA编码器的核心作用
3.2.1 消融实验结果
直接输入one-hot编码的U-Net模型性能显著降低,平均MCC仅0.07-0.11,远低于SegmentNT-3kb的0.37,证明DNA编码器的重要性。
随机初始化NT编码器的模型,平均MCC仅0.16,且收敛速度比预训练编码器模型慢7倍,表明基因组自监督预训练能大幅提升模型性能。
同时微调NT编码器和U-Net头的模型性能最优,且基于多物种预训练NT的SegmentNT优于人类基因组预训练NT模型(如图1e所示)。
3.2.2 与基线模型对比
原始SpliceAI架构(平均MCC 0.18)性能优于BPNet(0.10)、U-Net和随机初始化NT模型,缩放后SpliceAI平均MCC达0.27,但仍远低于SegmentNT-3kb的0.37(如图1e所示)。
3.3 SegmentNT对长序列的泛化能力
3.3.1 不同长度模型性能对比
随着训练序列长度增加(3kb→10kb→20kb→30kb),模型平均MCC持续提升,SegmentNT-30kb达到最高(0.45),蛋白质编码基因、3’UTR、外显子和内含子的性能改善尤为明显(如图2a、2b所示)。
3.3.2 上下文长度扩展效果
SegmentNT-10kb经上下文扩展后,在长序列上性能显著提升:100kb序列的平均MCC从0.07提升至0.26(如图2c所示)。
所有SegmentNT模型中,SegmentNT-30kb在各序列长度下均表现最佳,50kb序列输入时平均MCC达0.47,100kb时仍保持0.45的高值(如图2d所示)。
3.3.3 长序列注释示例
在含TMEM230/PCNA/CDS2三个重叠基因的50kb区域,SegmentNT-30kb准确预测了所有14种元件的位置和概率,单次输出70万个预测结果(14×50000)(如图2e所示)。
3.3.4 错误预测分析
所有元件的错误预测不仅集中在区域边缘,还富集于标记区域内部,调控元件的内部错误预测占比高于边缘,表明性能瓶颈源于部分区域的整体预测效果不佳,而非边缘效应。
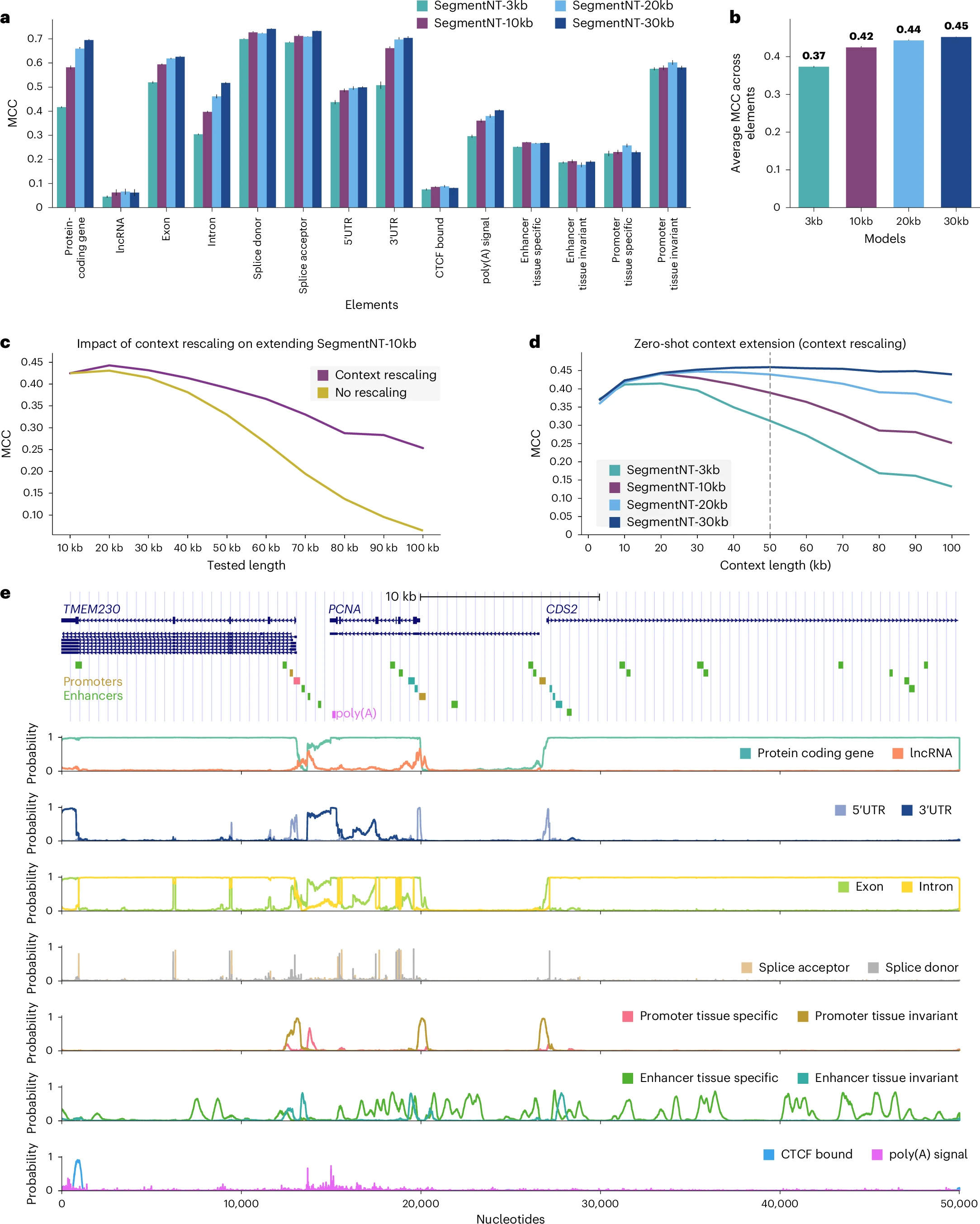
图注:a. 不同长度SegmentNT模型在14种元件上的MCC性能(数据为10次测试集采样的均值±95%置信区间);b. 各模型在14种元件上的平均MCC(数据为14种元件的均值±95%置信区间,每种元件经10次采样);c. 上下文扩展对SegmentNT-10kb在不同长度序列上的性能影响(平均MCC);d. 各SegmentNT模型在不同输入序列长度下的平均MCC(虚线为50kb最优推理长度);e. TMEM230/PCNA/CDS2基因位点50kb区域的14种元件注释及预测概率示例。
3.4 不同基础模型作为DNA编码器的性能对比
3.4.1 30kb输入序列下的性能
SegmentNT平均MCC(0.45)优于SegmentEnformer(0.34)和SegmentBorzoi(0.35)。SegmentNT在基因元件(蛋白质编码基因、UTR、外显子等)和短序列元件(剪接位点、多聚腺苷酸信号)上表现突出,而SegmentEnformer和SegmentBorzoi在长链非编码RNA、CTCF结合位点及调控元件上性能更优(如图3b、3c所示)。
3.4.2 扩展输入序列长度后的性能
SegmentEnformer(196kb输入)和SegmentBorzoi(524kb输入)相较于30kb版本性能整体提升,蛋白质编码基因、长链非编码RNA和内含子的改善最为显著。SegmentBorzoi在UTR区域的额外提升源于其RNA测序数据预训练(如图3b、3c所示)。
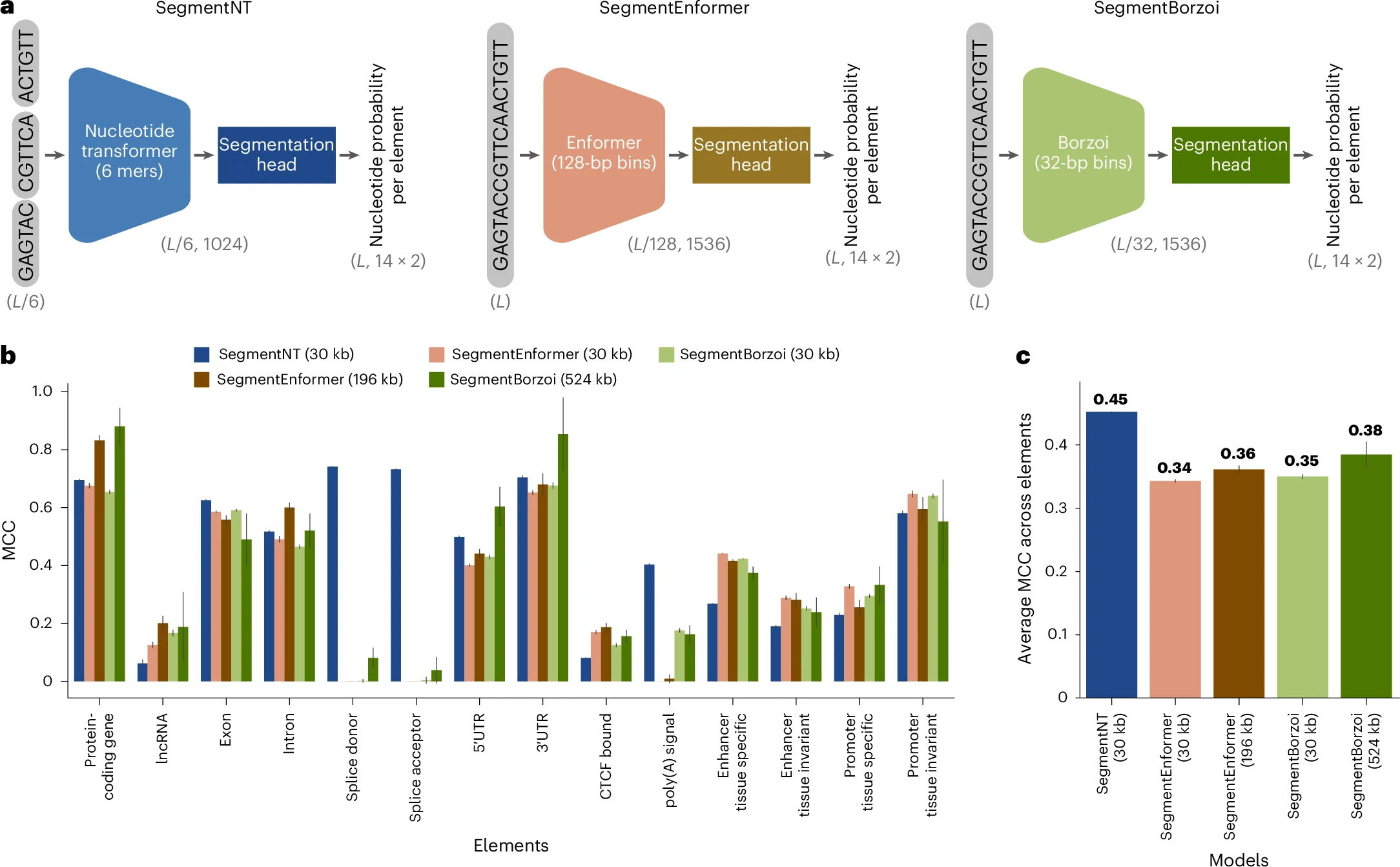
图注:a. SegmentNT、SegmentEnformer、SegmentBorzoi的架构示意图(括号内为输入输出维度,Enformer输出对应128bp bins,Borzoi对应32bp bins);b. 不同模型在14种元件上的MCC性能(数据为10次测试集采样的均值±95%置信区间);c. 各模型在14种元件上的平均MCC(数据为14种元件的均值±95%置信区间)。
3.5 与现有基因注释工具的对比
3.5.1 主异构体注释场景
SegmentNT-30kb在剪接供体位点上性能优于AUGUSTUS,内含子和剪接受体位点性能相当,编码序列(CDS)区域因精确率较低表现稍差(如图4a、4b所示)。
3.5.2 所有异构体注释场景
在30kb基因片段和全染色体测试集中,SegmentNT-30kb在所有基因元件上的F1分数和MCC均优于AUGUSTUS,且兼具更高的精确率和召回率(如图4c、4d、4e所示)。
3.5.3 区域水平评估
基于SOV分数的区域水平评估显示,SegmentNT-30kb在所有场景下仍优于AUGUSTUS,但全染色体测试集的性能提升幅度小于核苷酸水平指标(如图4f、4g、4h所示)。
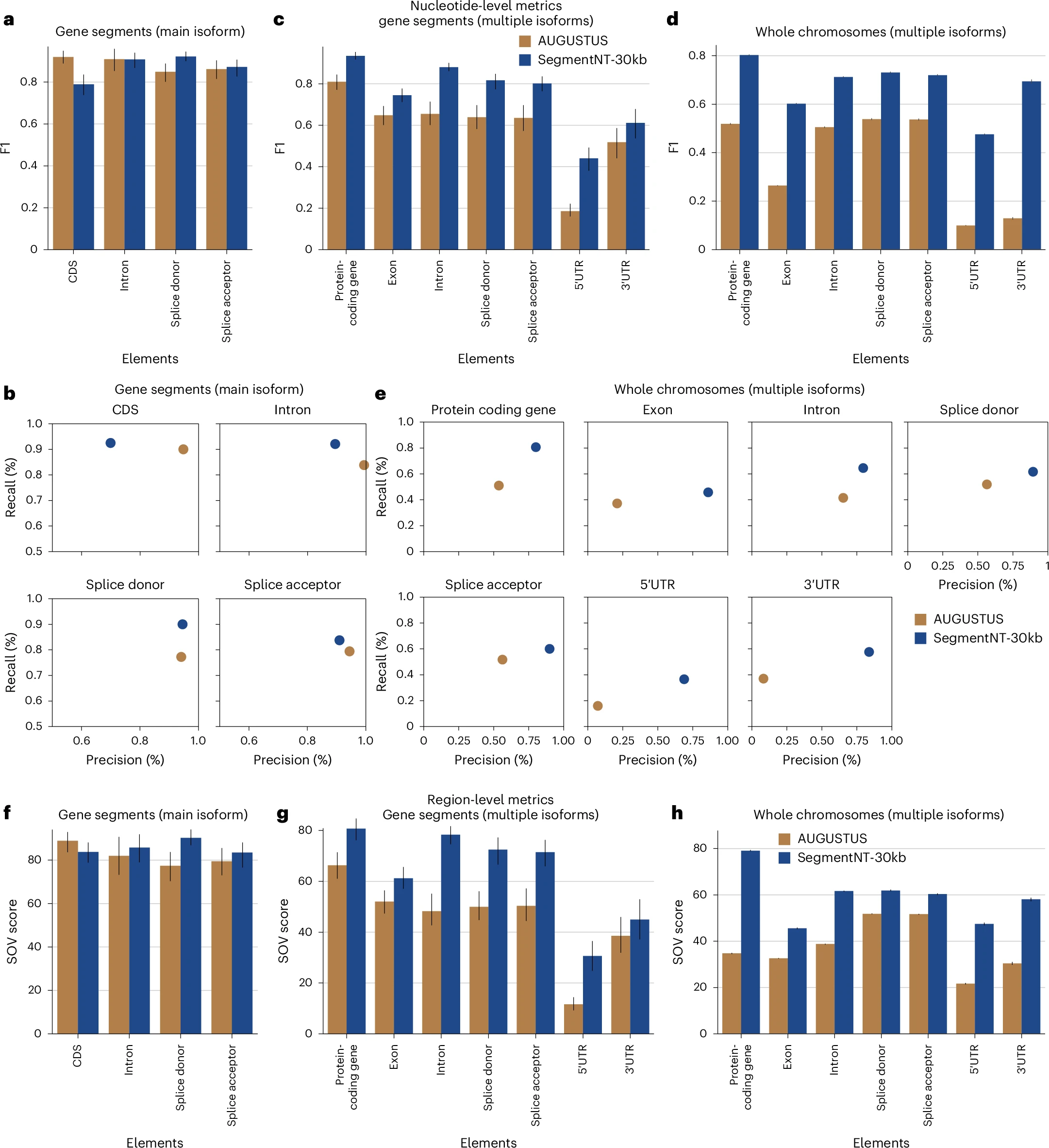
图注:a、c、d. 不同数据集(主异构体30kb片段、所有异构体30kb片段、所有异构体全染色体)中两种模型在各基因元件上的F1分数;b. 主异构体30kb片段数据集的精确率-召回率曲线;e. 所有异构体全染色体数据集的精确率-召回率曲线;f、g、h. 不同数据集下的SOV分数(误差线为95%置信区间,全染色体数据为10次采样均值±95%置信区间)。
3.6 剪接位点预测性能
3.6.1 基因位点示例
在EBF4基因位点,SegmentNT-30kb准确预测了所有外显子、内含子及剪接位点(包括起始处的可变外显子),而SpliceAI和Pangolin存在转录本边界外的假阳性预测(如图5a所示)。
3.6.2 mRNA测试集性能
SegmentNT-30kb与SpliceAI、Pangolin性能相当:剪接供体位点auPRC均为0.93-0.94,剪接受体位点auPRC为0.93-0.96(如图5b、5c、5d所示)。
3.6.3 全染色体测试集性能
SegmentNT-30kb的MCC高于SpliceAI和Pangolin,auPRC表现相当(剪接供体0.68-0.74,剪接受体0.70-0.72),剪接供体位点的top-k准确率更优(如图5e、5f、5g所示)。
3.6.4 非编码RNA剪接位点预测
SegmentNT和Pangolin在非编码RNA剪接位点上的性能低于SpliceAI,提示编码序列的相关信号可能驱动剪接检测性能(补充图6c)。
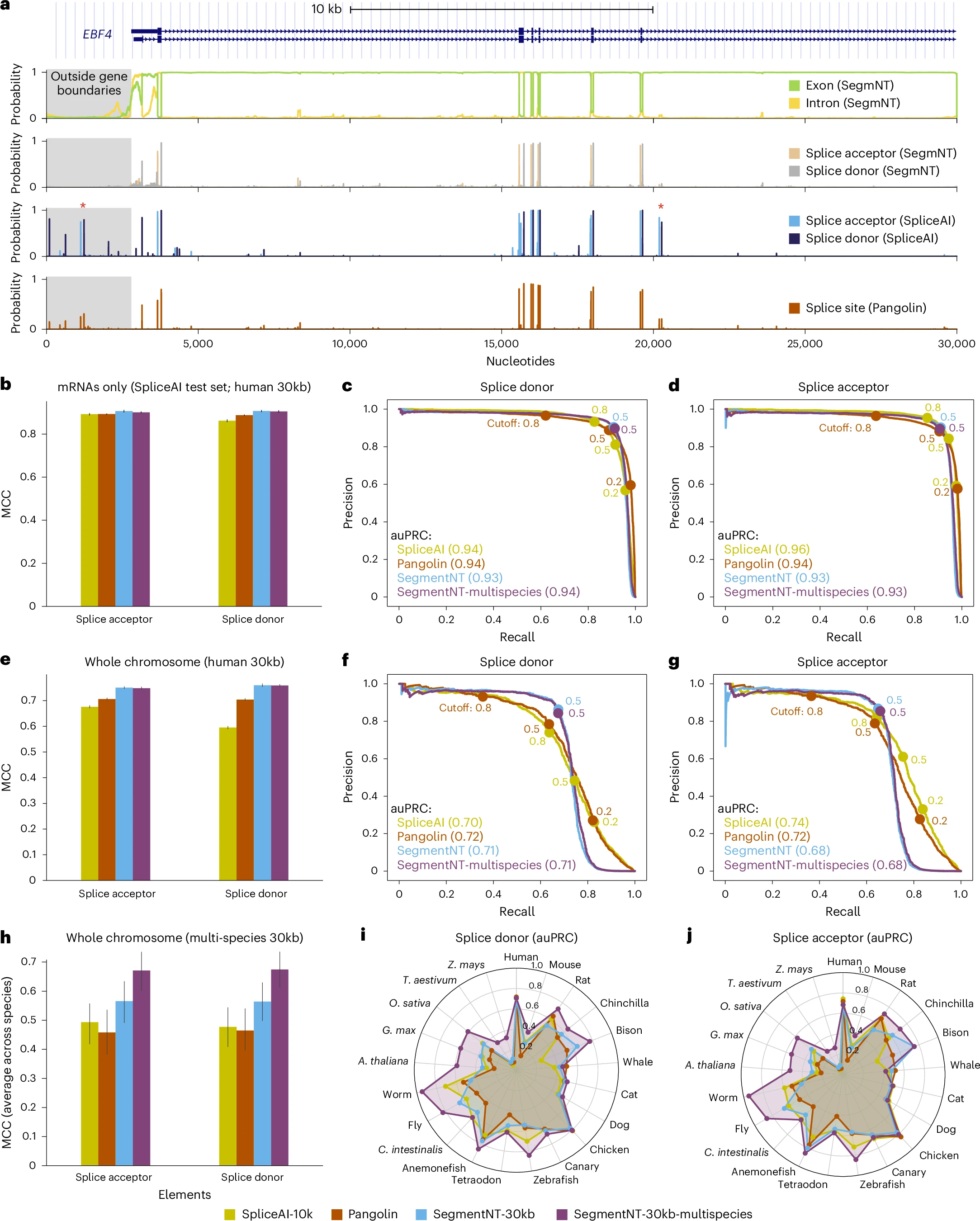
图注:a. EBF4基因位点的剪接元件预测示例(红色星号标记SpliceAI/Pangolin的错误预测区域);b. mRNA测试集上的MCC性能(误差线为100次自助抽样的95%置信区间);c、d. mRNA测试集的精确率-召回率曲线及auPRC值;e. 人类全染色体测试集的MCC性能;f、g. 人类全染色体测试集的精确率-召回率曲线及auPRC值;h. 多物种全染色体测试集的平均MCC;i、j. 多物种剪接供体/受体位点的auPRC雷达图。
3.7 调控元件定位性能
SegmentNT-30kb在启动子和增强子注释上优于滑动窗口基线模型,SegmentEnformer表现最佳(补充图7b、7c)。DeePromoter在 curated测试集上性能优异,但在基因组序列语境中泛化能力差于滑动窗口NT模型。
SegmentNT家族模型的推理速度更快,单次完成所有核苷酸预测,适用于遗传变异候选区域和个性化基因组的快速评估(补充图7d)。
3.8 SegmentNT的跨物种泛化能力
3.8.1 人类模型的跨物种表现
人类SegmentNT-30kb模型在不同物种中均表现出高性能,外显子和剪接位点的MCC最高(与进化保守性相关)。亲缘关系较近的物种(如大猩猩、猕猴)性能较好,进化距离较远的动物和植物性能下降(如图6b、6c所示)。
3.8.2 物种进化距离与性能关联
随着与人类的分化时间增加,各基因元件的MCC呈下降趋势,外显子和剪接位点的下降幅度最小(如图6c所示)。
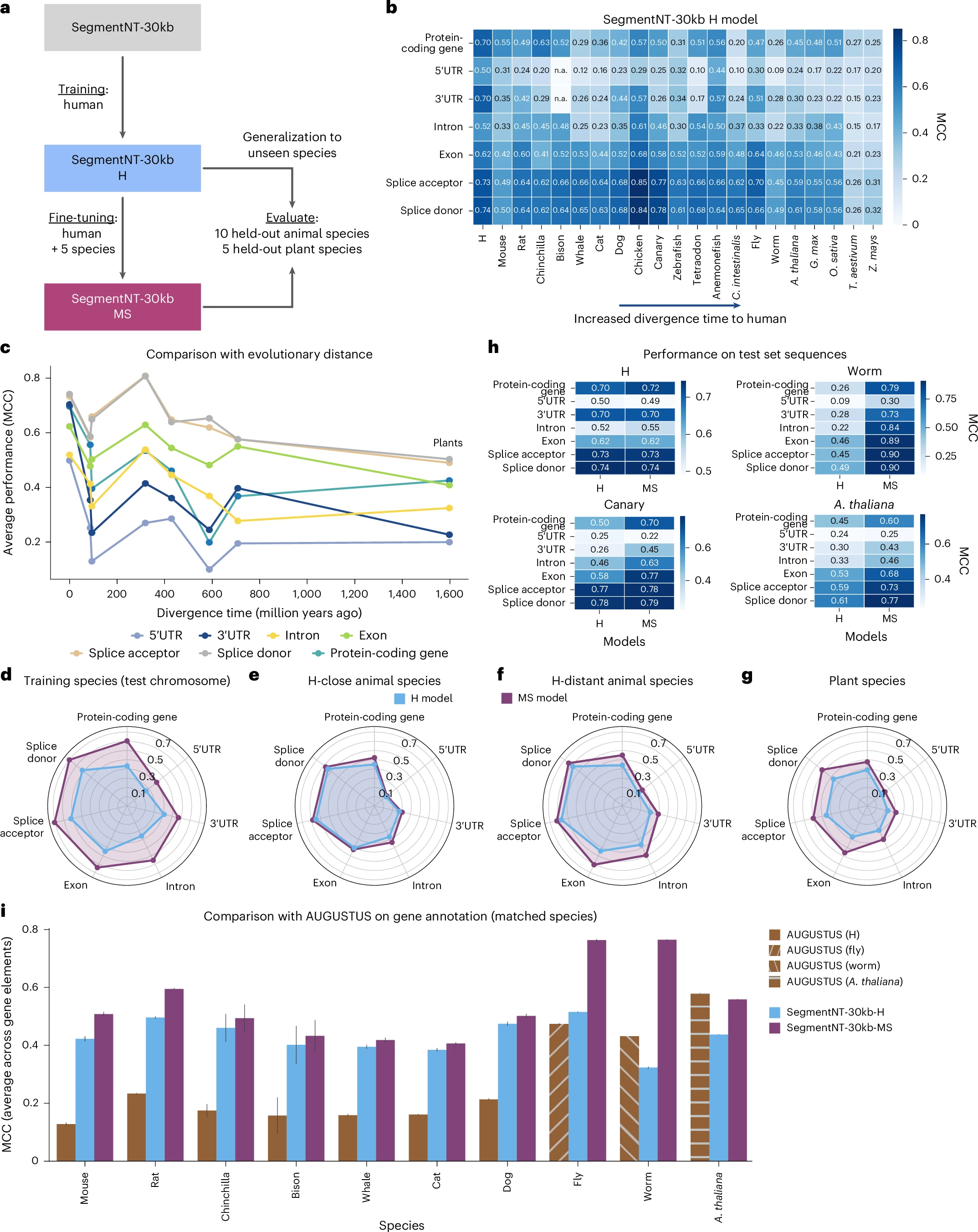
图注:a. 多物种模型微调与跨物种评估示意图;b. 人类模型在各物种基因元件上的MCC性能(按与人类的分化时间排序);c. 基因元件MCC与进化分化时间的关联(相同进化距离物种的MCC均值);d-g. 人类模型与多物种模型在训练物种、近缘动物、远缘动物、植物上的性能雷达图;h. 四种代表性物种的各元件MCC对比;i. 与AUGUSTUS在不同物种基因注释上的平均MCC对比(误差线为95%置信区间)。
3.9 多物种SegmentNT模型的泛化提升
3.9.1 训练物种性能
多物种模型在训练物种的测试染色体上性能优于人类模型(如图6d所示)。
3.9.2 未见过物种的泛化
- 人类近缘动物(分化时间<1000万年):两模型性能相当(平均MCC 0.62 vs 0.64)
- 人类远缘动物(分化时间>1000万年):多物种模型平均MCC从0.49提升至0.57
- 植物物种:多物种模型平均MCC从0.34提升至0.45,即使未经过植物基因组训练仍表现出显著改善(如图6e、6f、6g所示)。
3.9.3 与AUGUSTUS的对比
多物种模型在除拟南芥外的所有物种上,基因注释性能均优于AUGUSTUS(如图6i所示)。
4 讨论
4.1 核心结论
- 提出的DNA基础模型微调方法,实现了14种基因组元件的单核苷酸分辨率注释,SegmentNT模型可处理长达50kb的DNA序列,单次输出70万个预测结果,效率显著。
- 预训练DNA编码器是模型高性能的关键,相较于直接使用one-hot编码或随机初始化编码器,性能提升显著,且上下文长度扩展方法有效突破了序列长度限制。
- 不同DNA编码器各具优势:SegmentNT在基因元件和短序列元件上表现最佳,SegmentEnformer和SegmentBorzoi在调控元件上更具优势,可根据任务需求选择。
- 人类SegmentNT模型具有天然的跨物种泛化能力,多物种模型进一步提升了对远缘动物和植物的注释性能,为未充分研究物种的基因组注释提供了高效工具。
4.2 研究展望
- 扩展SegmentNT的上下文长度,结合自然语言处理领域的长序列建模技术和状态空间模型,开发下一代模型。
- 细化调控元件分类,按细胞类型拆分启动子和增强子,提升细胞类型特异性调控密码的预测精度。
- 探索SegmentNT在遗传变异影响评估、癌症基因组分析等领域的应用,整合实验数据进一步优化注释流程。
- 扩充多物种模型的训练物种范围,纳入更多植物物种及基因组重排显著的物种,提升序列多样性覆盖。
数据获取说明
SegmentNT训练数据来源于公开资源。基因注释数据来自gencode(https://www.gencodegenes.org/)和Ensembl数据库(https:// www.ensembl.org)。人类调控元件数据来自encode的筛选数据库(https://screen.wenglab.org/)。 进化距离数据取自生命时间树(Timetree of Life)。SpliceAI测试集数据源自Illumina Basespace平台(https://basespace.illumina.com/projects/66029966/)。 交互式浏览器会话,展示人类SegmentNT-30kb模型在测试染色体20和21上位于 https://tinyurl.com/23837bnl 区域的标签与预测结果。
代码资源说明
人类及多物种SegmentNT-30kb模型、SegmentEnformer和SegmentBorzoi模型的模型权重,以及Jax语言的推理代码,可通过GitHub(https://github.com/instadeepai/nucleotide-transformer?tab=readme-ov-file#-segmentnt--family-segmentenformer-segmentborzoi)获取研究使用。HuggingFace平台上的PyTorch版本模型可访问(https:// huggingface.co/collections/InstaDeepAI/segmentnt-65eb4941c57 808b4a3fe1319)。示例笔记本可在Google Colab获取。
参考文献
- [1] de Almeida, B.P., Dalla-Torre, H., Richard, G. et al. Annotating the genome at single-nucleotide resolution with DNA foundation models. Nat Methods (2025). https://doi.org/10.1038/s41592-025-02881-2
加关注
关注公众号“生信之巅”。
 |
 |



