
2025年6月,香港科技大学计算机科学与工程系的Yihui Wang、Zhiyuan Cai及Hao Chen(通讯作者)等研究者在bioRxiv预印本发表了题为“Genomic Touchstone: Benchmarking Genomic Language Models in the Context of the Central Dogma”的学术文章。该文章报道了其开发的“Genomic Touchstone”综合基准测试框架,在基因组语言模型(gLMs)评估领域具有建立统一评估标准、指导模型设计与实际应用选择的重要意义。下面我将对文章进行解读。
1 摘要
基因组语言模型(gLMs)的出现革新了基因组序列分析,但现有评估缺乏覆盖中心法则全链条的整体框架。本研究提出Genomic Touchstone基准测试框架,涵盖36个任务、88个数据集,包含53.4亿碱基对的基因组序列,跨越DNA、RNA、蛋白质三种分子模态。通过评估34种代表性模型(含Transformer、CNN及Hyena、Mamba等高效架构),得出四大核心发现:(1) gLMs在RNA和蛋白质任务上表现比肩甚至优于专门预训练的模型;(2) Transformer模型整体领先,但高效序列模型在特定任务中潜力显著;(3) gLMs的缩放规律尚未完全明确,更长输入序列和多样化预训练数据持续提升性能,但模型规模增大未必带来更好结果;(4) 预训练策略(训练目标、语料组成)对下游泛化能力影响显著。该框架为人类基因组学领域的gLMs评估提供了统一标准,为模型设计和泛化能力研究奠定基础。
2 引言
2.1 研究背景
大型语言模型(LLMs)已革新自然语言处理,受基因组生物代码与人类语言的相似性启发,研究者将LLM框架扩展至生物序列分析,AlphaFold、ESM、DNABERT等模型的成功证明了其潜力。gLMs的设计围绕DNA序列的统计和功能特性展开,但现有评估存在诸多局限:任务覆盖有限,多聚焦分类问题且偏离真实生物分布;缺乏跨中心法则的全面评估,大多仅关注DNA任务;评估协议缺乏区分度,难以指导模型选择。
2.2 研究目标
开发一个基于中心法则、一致且具有区分度的综合基准测试框架Genomic Touchstone,系统评估gLMs在DNA、RNA、蛋白质多模态任务中的性能,为生物研究者提供模型选择参考,同时指导未来gLMs的设计与优化。
3 研究结果
3.1 Genomic Touchstone框架概述
基因组基准框架(Genomic Touchstone)概述
基因组基准框架(Genomic Touchstone)是一个全面的基准测试框架,旨在对基因组语言模型(gLMs)应对以人为中心的实际生物学挑战的能力进行全面且公正的评估。受中心法则启发——该法则描述了遗传信息从DNA到RNA再到蛋白质的顺畅流动,Genomic Touchstone是首个涵盖跨越DNA、RNA和蛋白质序列模态任务的基准测试(图1),用于严格评估当前基因组语言模型的性能与通用性。
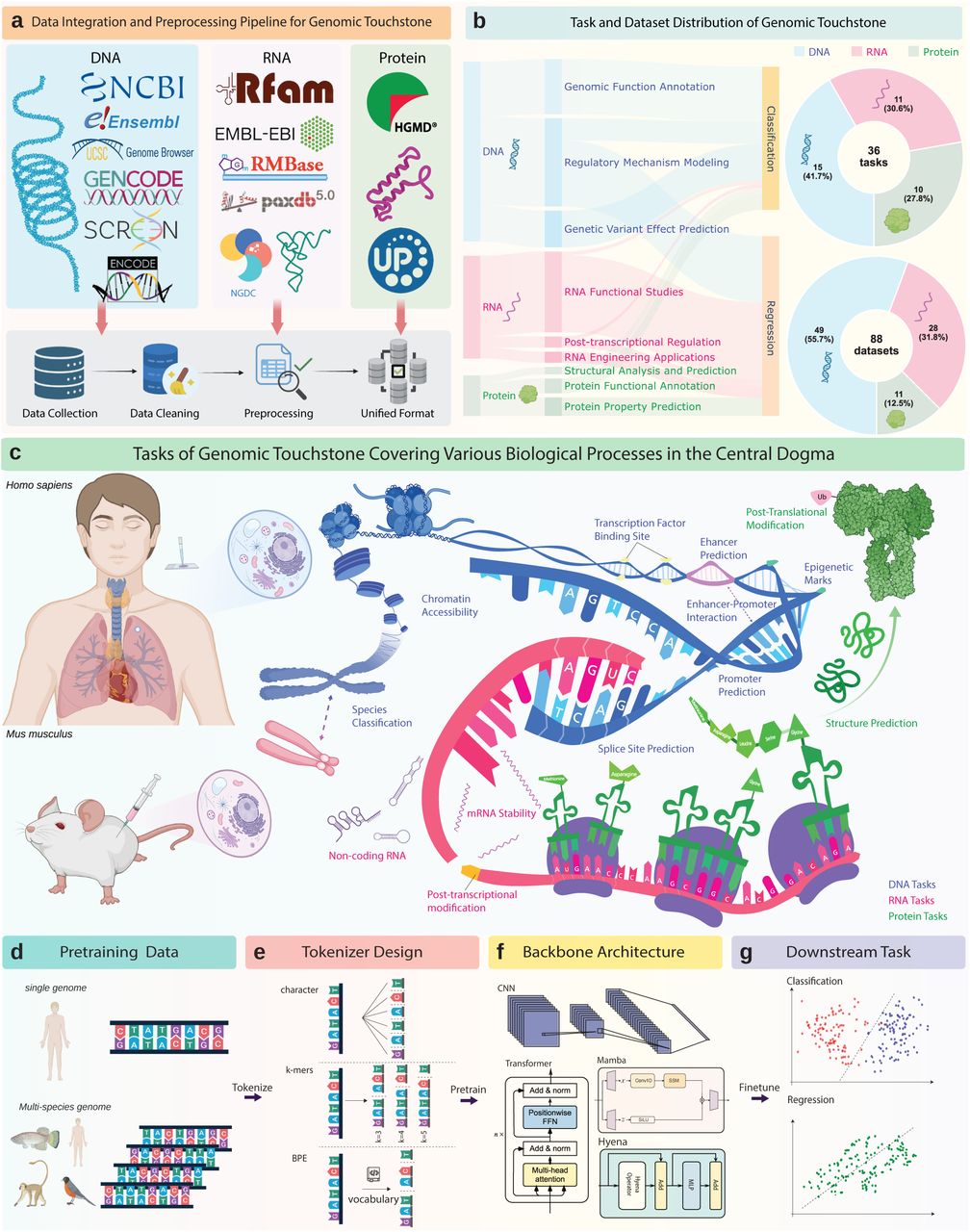
a:数据整合流程,从组学数据库收集原始资源,经清洗后将DNA、RNA和蛋白质序列转换为统一格式;b:基准测试组成,含36个任务和88个数据集,左侧为9大任务类别,右侧为各模态任务分布;c:基准测试任务覆盖的中心法则核心流程;d-g:基准测试框架 pipeline,包括预训练数据组成、分词策略、模型架构和下游任务分组。
基因组层面的核心挑战
这些挑战的核心在于基因组本身的复杂性。DNA是生命的基本分子,承载着细胞功能、发育和进化所需的遗传指令。它包含蛋白质编码基因、非编码RNA以及各种调控基序等多种元件(图1c)。基因组功能注释致力于识别和分类这些元件,以揭示它们在细胞过程中的作用。性能优异的基因组语言模型能够利用从大型基因组数据集获得的知识,检测复杂的序列模式。这种能力可绘制出全面的功能元件图谱,对推进理论基因组学发展和疾病诊断等实际应用都至关重要。
基因表达受包含启动子、增强子和其他元件的复杂调控网络控制。准确建模这些机制需要基因组语言模型捕捉长程依赖关系和上下文特异性相互作用,这对于理解基因在不同细胞环境中如何被激活或沉默至关重要。同时,DNA序列中的变异(即使是单核苷酸层面的变异)也可能对基因功能产生深远影响并引发疾病,因此预测这些遗传变异的影响,对于理解发病机制和开发靶向治疗具有重要意义。
在该领域表现出色的基因组语言模型应能分析突变带来的细微差异,揭示其对基因调控和表型的潜在影响,进而提供关键的预测能力,助力将计算基因组学的见解转化为可行的临床策略。因此,我们对基因组语言模型在DNA层面的评估聚焦于三个类别:基因组功能注释、调控机制建模和遗传变异影响预测(图1b)。这三个类别共包含15项任务,旨在考验模型准确解读和阐释基因组信息多方面特征的能力。
RNA层面的任务设计
与主要作为遗传信息储存库的DNA不同,RNA主动将这些指令转化为功能蛋白,调控基因表达并调节细胞对内外刺激的反应(图1c)。作为DNA和蛋白质之间的关键中介,RNA在蛋白质合成、酶活性和基因调控等众多生物学过程中发挥着核心作用。因此,揭示RNA的多样功能,对于理解细胞复杂性和疾病潜在机制至关重要。
从语言学角度看,RNA和DNA的“词汇表”仅相差一个“字母”:RNA中的尿嘧啶(U)取代了DNA中的胸腺嘧啶(T)。但尿嘧啶和胸腺嘧啶均为与腺嘌呤配对的嘧啶类碱基,结构相似,这表明它们功能相近。基于这种相似性,我们推测RNA和DNA可能处于统一的语义空间中。这意味着基于DNA预训练的基因组语言模型,或许能有效泛化到RNA相关任务。
受BEACON的启发,我们收集了三类主要的RNA任务:RNA功能研究、转录后调控及RNA工程应用(图1b)。RNA功能研究分析RNA如何调控基因表达并参与疾病发生,为治疗干预提供机制层面的见解;转录后调控聚焦转录后发生的RNA加工、稳定性调控及修饰事件,这些过程共同决定RNA的寿命和调控效果;RNA工程应用则探索RNA在合成生物学中的潜力,以提升其在生物技术和医学领域的应用价值,应对复杂的生物学挑战。
蛋白质层面的评估逻辑
通过分子生物学中心法则,蛋白质与DNA紧密相连——该法则描述了遗传信息从DNA到RNA,最终到蛋白质的传递过程。在这一过程中,DNA作为稳定的遗传指令储存库,先转录为信使RNA(mRNA),再翻译为蛋白质。而蛋白质则执行从催化、结构支撑到信号传导和调控的关键细胞功能。这种顺畅的信息传递支撑着所有生物学过程,也体现了基因组序列与其编码的蛋白质组之间的内在联系(图1c)。
由于基因组(尤其是编码DNA序列CDS)编码了所有蛋白质,基于基因组数据预训练的基因组语言模型不仅能预测DNA序列特征,还能预测蛋白质特性。为评估基因组语言模型处理蛋白质相关任务的能力,我们筛选了一系列涵盖蛋白质生物学多个方面的任务。通过提取每种蛋白质对应的编码DNA序列(CDS),将蛋白质水平的数据集转换为基于DNA的格式。序列获取的具体流程详见方法部分。
具体而言,我们的任务套件包含三个不同类别(图1b):首先是结构分析与预测,评估基因组语言模型直接从序列数据中推断蛋白质二级和三级结构的能力;其次是蛋白质功能注释,考察模型根据蛋白质的序列特征识别并赋予其生物学功能的效果;最后是蛋白质特性预测,聚焦于预测稳定性、溶解性和结合亲和力等关键理化属性,以此展现模型预测序列变异对蛋白质功能影响的能力。
模型选择与评估设计
基于精心筛选的数据集,我们选取了多种基因组语言模型,以及两种基准模型(卷积神经网络CNN和长短期记忆网络LSTM),评估它们在与人类相关任务中的性能。为确保评估全面且具有生物学意义,我们重点关注基于人类基因组数据预训练的模型——包括仅在人类参考基因组上训练的模型,以及使用包含大量人类序列的多物种语料库训练的模型(详见方法部分)。
所选模型涵盖多种架构,从传统的CNN、LSTM,到基于Transformer的模型,再到BigBird、Hyena和Mamba等新兴架构,每种模型均搭配不同的分词策略(如字符级、k-mer和字节对编码BPE)。模型规模从数百万参数的小型架构到高达25亿参数的大规模模型不等,使我们能够探究模型容量对生物序列建模的影响。
这一系列基于人类数据预训练的模型,让我们得以系统评估架构选择、分词方案和预训练数据对下游人类功能基因组学任务性能的影响。通过在统一的基准测试集(涵盖DNA、RNA和蛋白质层面的任务)上对所有模型进行微调,我们分离出架构设计、预训练策略和参数规模等特定建模因素的影响。纳入RNA和蛋白质语言模型,进一步使我们能够探索这些模型跨分子模态的泛化能力。
值得注意的是,没有任何单一模型在所有任务中持续优于其他模型(图2a);即使是表现最佳的模型,平均排名也仅为7.3,这凸显了当前基因组模型固有的权衡取舍和专门化特性。这种差异性强调了多维度评估的重要性,也表明不同任务可能受益于不同的归纳偏好。最后,我们扩展分析以验证大语言模型(LLM)中观察到的缩放定律是否适用于基因组领域,探究模型容量和数据构成如何共同影响在健康相关及更广泛生物学场景中的泛化能力。
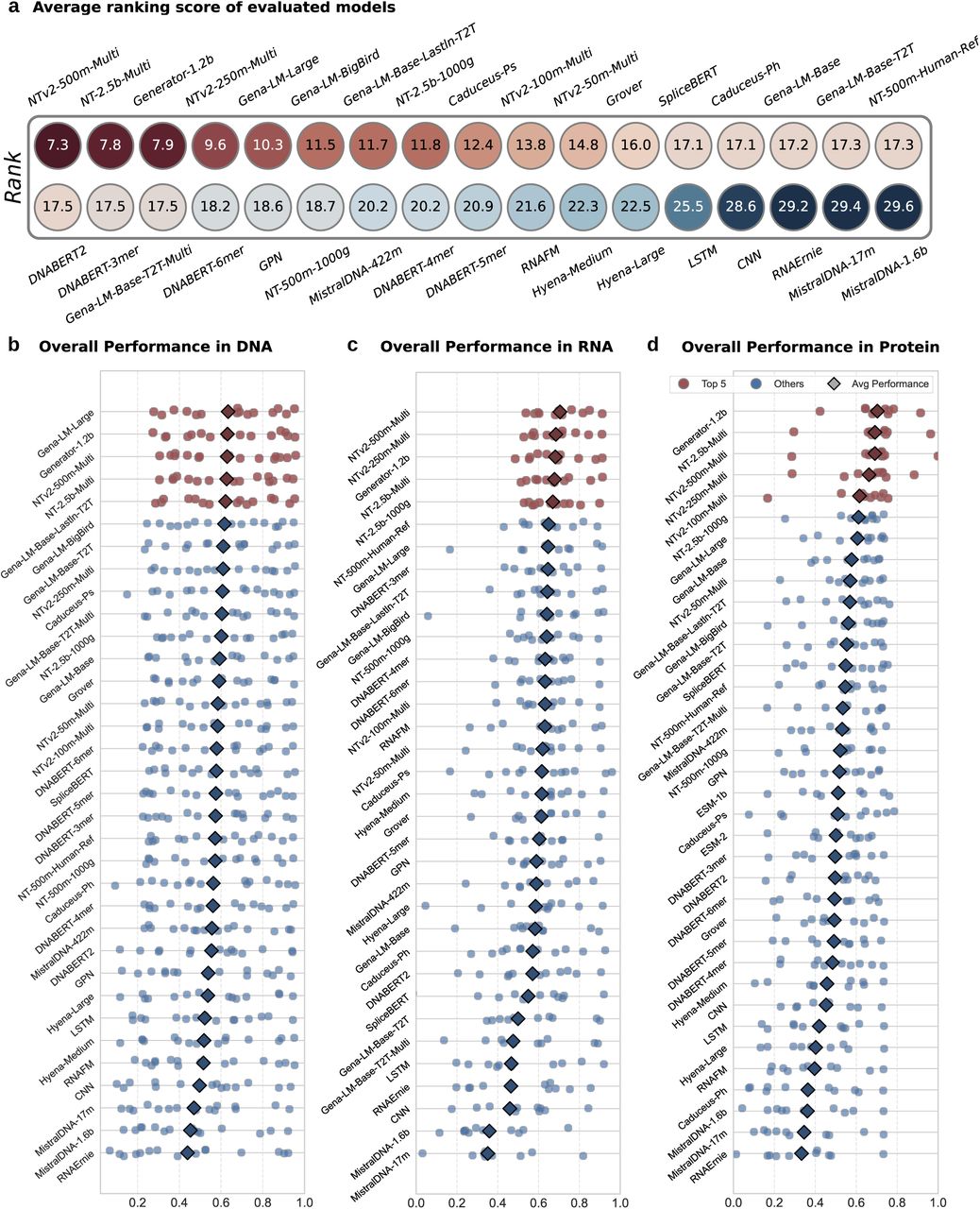
a. 基于整体性能的模型平均排名。每个圆圈代表一个模型,颜色深浅对应排名(颜色越深表示性能越优),中心位置显示数字排名。b-d. 各模型在DNA(b)、RNA(c)和蛋白质(d)基准测试中的任务特定性能分布(点状图)与平均性能分布(黑色菱形)。各模态中排名前五的模型以红色高亮显示,其余模型则以蓝色标注。
3.2 DNA相关任务性能
DNA任务分为基因组功能注释、调控机制建模、遗传变异效应预测三类,核心结果如下:
- 基因组功能注释:
剪接位点预测(SpliceBERT的F1分数0.918)和CpG甲基化预测(NTv2-500m-Multi的F1分数0.957)任务表现优异;表观遗传标记预测难度最高(顶级模型F1分数仅0.380);输入序列长度提升显著改善物种分类等任务性能,长上下文模型(如Hyena-Large)在8192碱基对以上序列中表现突出。 - 调控机制建模:
增强子-启动子相互作用预测表现较好(DNABERT2的F1分数0.941);基因表达预测(Generator-1.2b的$R^2$=0.548)和增强子活性预测(NT-2.5b-1000g的Spearman分数0.317)性能中等,模型差异显著。 - 遗传变异效应预测:
BRCA1/2 SNV功能影响分类任务表现最佳(NT-2.5b-1000g的F1分数0.893);致病性分类整体难度高(顶级模型F1分数仅0.391);GPN在遗传疾病分类中领先(F1分数0.725)。
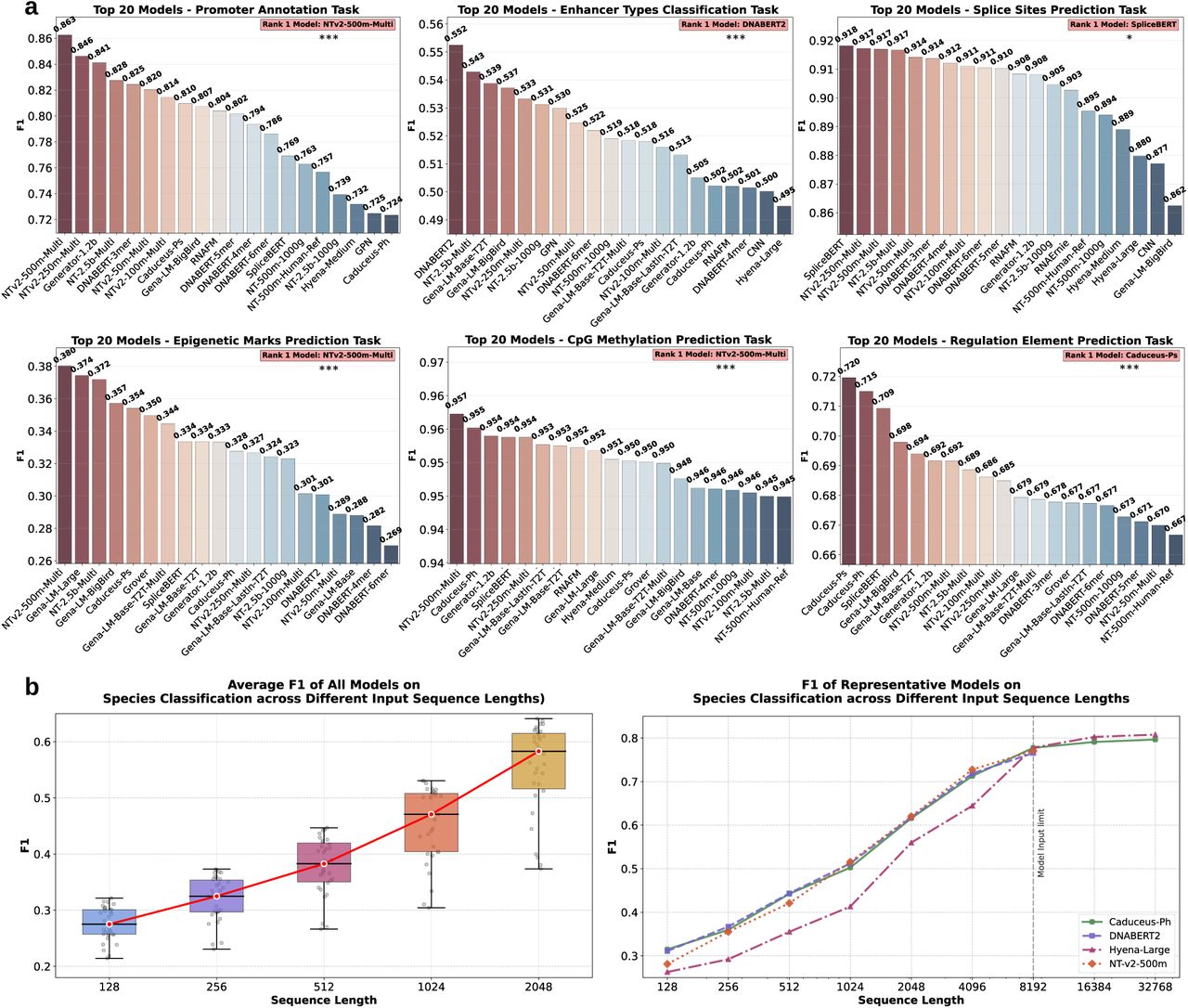
a:6项代表性基因组功能注释基准测试中前20个模型的F1分数(***: P<0.001, *: P<0.05),不同模型在不同任务中表现各异;b:不同输入序列长度下的物种分类性能,左图为多个模型在不同序列长度下的平均F1分数分布及趋势线,右图显示长序列长度显著提升F1分数,长上下文模型在8192碱基对以上序列中独占优势。
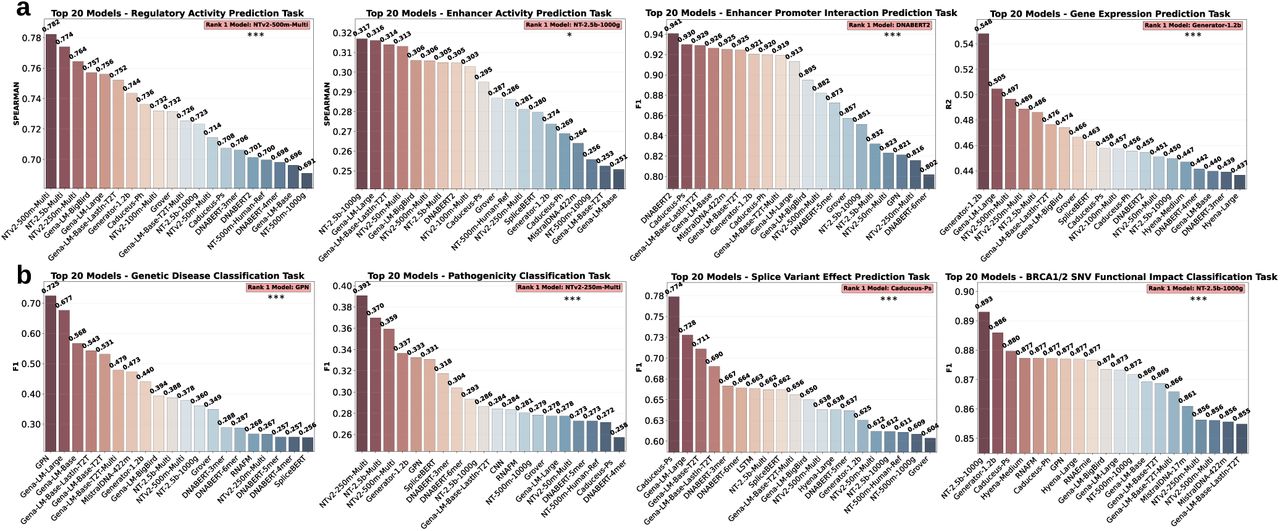
a. 调控机制建模任务的前20个模型,涵盖调控活性预测、增强子活性预测、增强子-启动子相互作用预测及基因表达预测(***:P < 0.001)。b. 基因变异效应预测任务的表现,包括遗传疾病分类、致病性分类、剪接变异效应预测以及BRCA1/2 SNV 功能影响分类(***:P < 0.001)。
3.3 RNA相关任务性能
RNA任务分为RNA功能研究、转录后调控、RNA工程应用三类,核心发现:
- DNA预训练gLMs在几乎所有RNA任务中优于RNA预训练模型,即使参数规模相当(如DNABERT2、NTv2-100m-Multi vs RNAErnie、RNAFM)。
非编码RNA分类(Caduceus-Ps的F1分数0.960)和APA异构体预测(Caduceus-Ps的Spearman分数0.929)表现突出;平均核糖体负载预测(Hyena-Medium的Spearman分数0.812)和mRNA稳定性预测(Generator-1.2b的Spearman分数0.554)呈现模型差异。
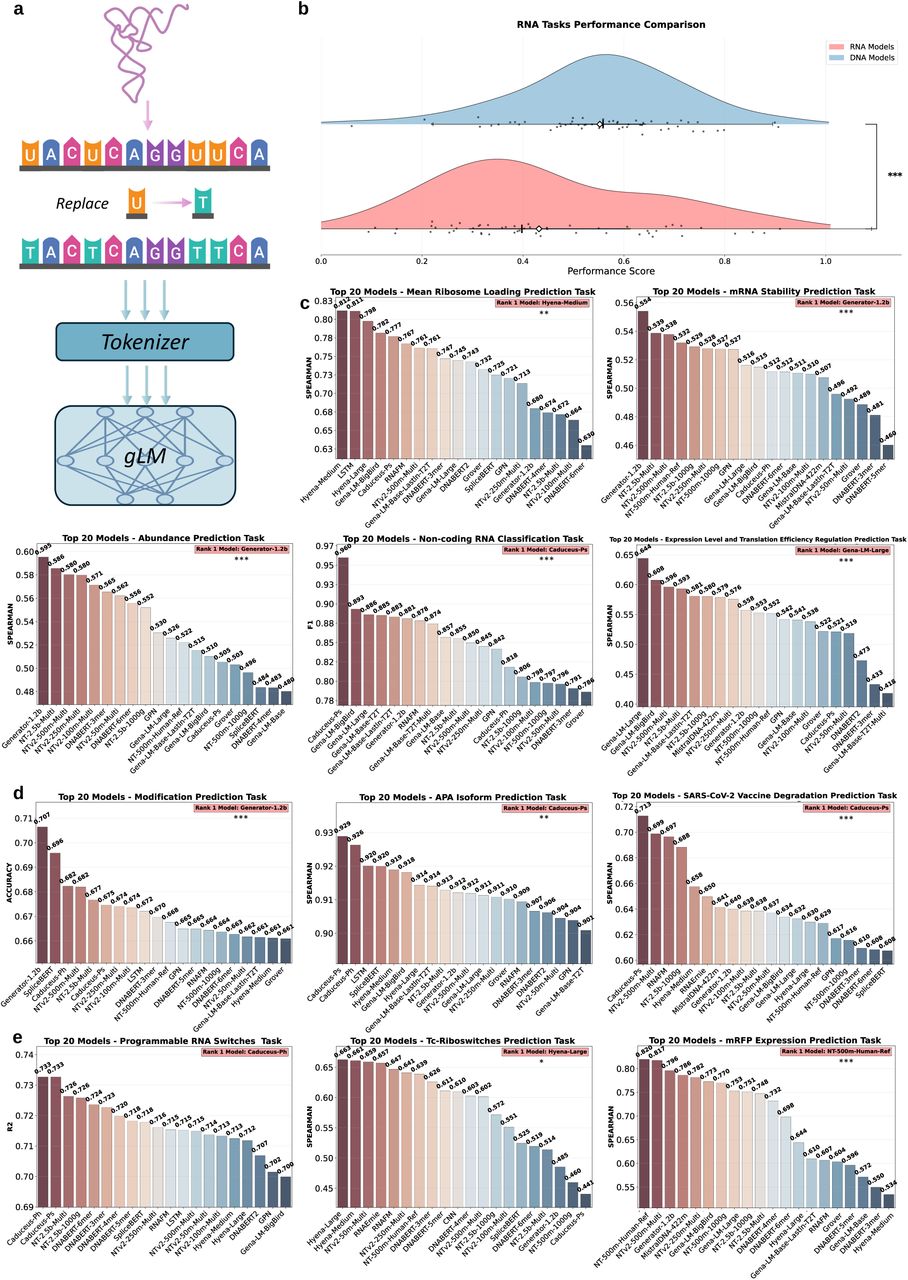
a:RNA序列转换为DNA输入的示意图,尿嘧啶(U)替换为胸腺嘧啶(T)后进行分词和模型输入;b:RNA预训练与DNA预训练模型在RNA任务上的性能对比,DNA模型(蓝色)显著优于RNA模型(红色)(***: P < 0.001);c-e:不同RNA任务类别中前20个模型的性能,包括RNA功能研究(c)、转录后调控(d)和RNA工程应用(e)(***: P<0.001, **: P < 0.01, *: P < 0.05)。
3.4 蛋白质相关任务性能
蛋白质任务分为结构分析与预测、功能注释、性质预测三类,核心结果:
- DNA预训练gLMs在多数蛋白质任务中优于蛋白质预训练模型(如NTv2-500m-Multi在7/10项任务中超越ESM-2)。
- 蛋白质
结构域分类(NTv2-500m-Multi的F1分数1.0)和跨膜区域预测(Generator-1.2b的F1分数0.782)表现优异;酶分类任务整体难度较高(Generator-1.2b的F1分数0.423);蛋白质稳定性预测中ESM-1b领先(Spearman分数0.762)。
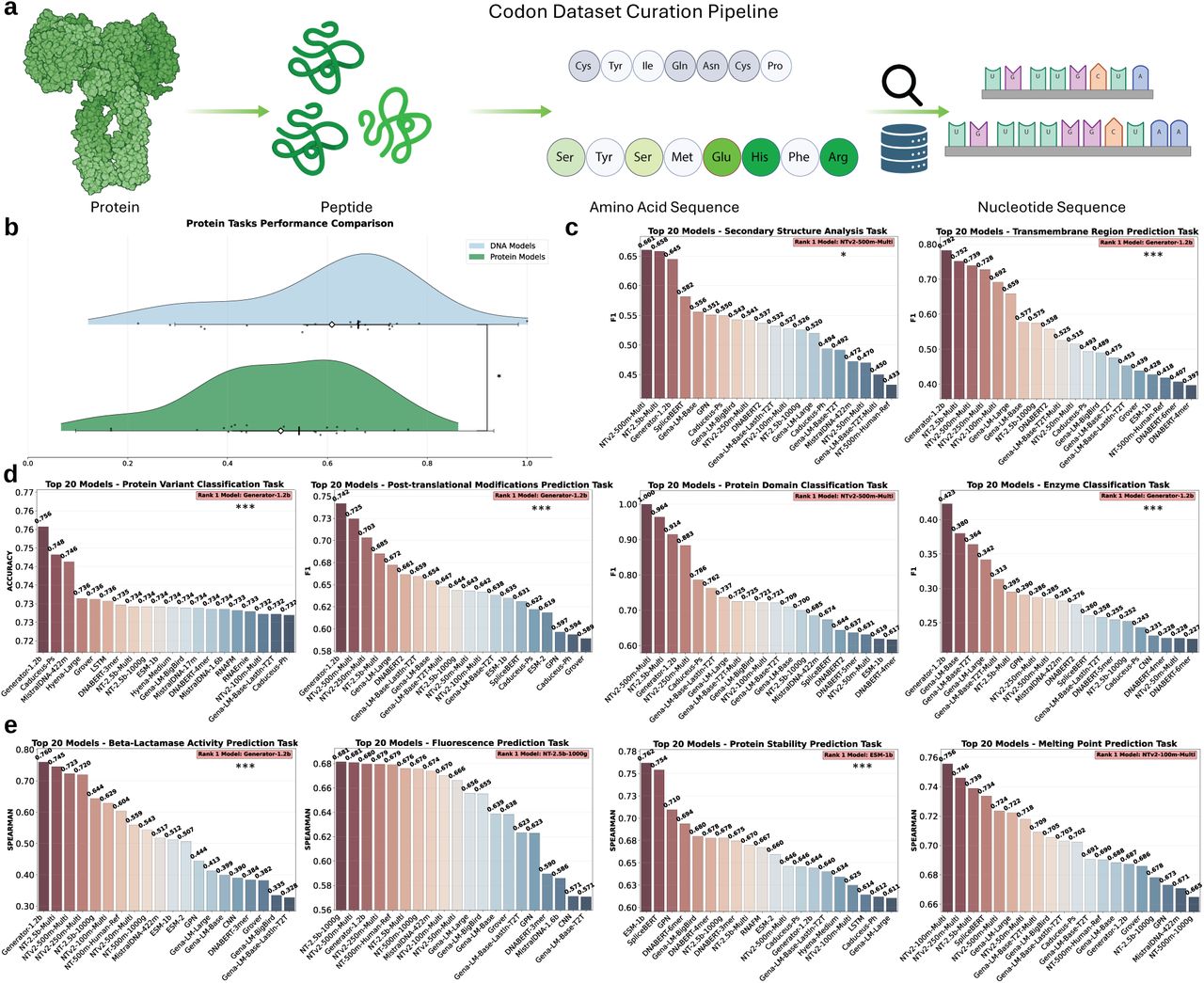
a. 密集式蛋白质层面基准测试模型性能。密码子数据集构建流程:蛋白质分子经计算机模拟消化为多肽,翻译为氨基酸序列,再逆向翻译为核苷酸(密码子)序列,经过过滤和注释后生成任务就绪数据集。b. 蛋白质与DNA预训练模型在蛋白质任务上的性能对比。DNA模型(蓝色)显著优于蛋白质模型(绿色),统计学显著性高(*P<0.05)。为确保公平比较,我们选取了参数量相近的模型:DNA模型采用NTv2-500m-Multi和NT-500m-1000G,蛋白质模型采用 ESM -1b和 ESM -2——所有模型参数量均约5亿。c. 结构分析与预测任务前20名模型表现,包括二级结构分析和跨膜区域预测(***:P<0.001,*:P<0.05)。d. 蛋白质功能注释任务前20名模型表现,涵盖蛋白质变体分类、翻译后修饰预测、蛋白质结构域分类及酶分类(***:P<0.001)。e. 蛋白质性质预测任务前20名模型表现,包括β-内酰胺酶活性预测、荧光预测、蛋白质稳定性预测及熔点预测(***:P<0.001)。
3.5 模型架构、缩放规律与预训练策略洞察
- 架构影响:Transformer 模型占据前15名中的14个,整体性能领先,但SSM-based模型(如Caduceus、Hyena)在长序列任务中具有潜力,线性时间复杂度优势显著。
- 缩放规律:NT系列模型呈现明确缩放趋势(参数增加、预训练数据多样化提升性能),但其他模型未表现出一致规律,短序列预训练限制了模型潜力发挥。
- 预训练策略:多物种预训练数据(如NT-2.5b-Multi)、长输入序列预训练显著提升模型泛化能力;融入生物先验的预训练目标(如Generator模型)带来性能增益。
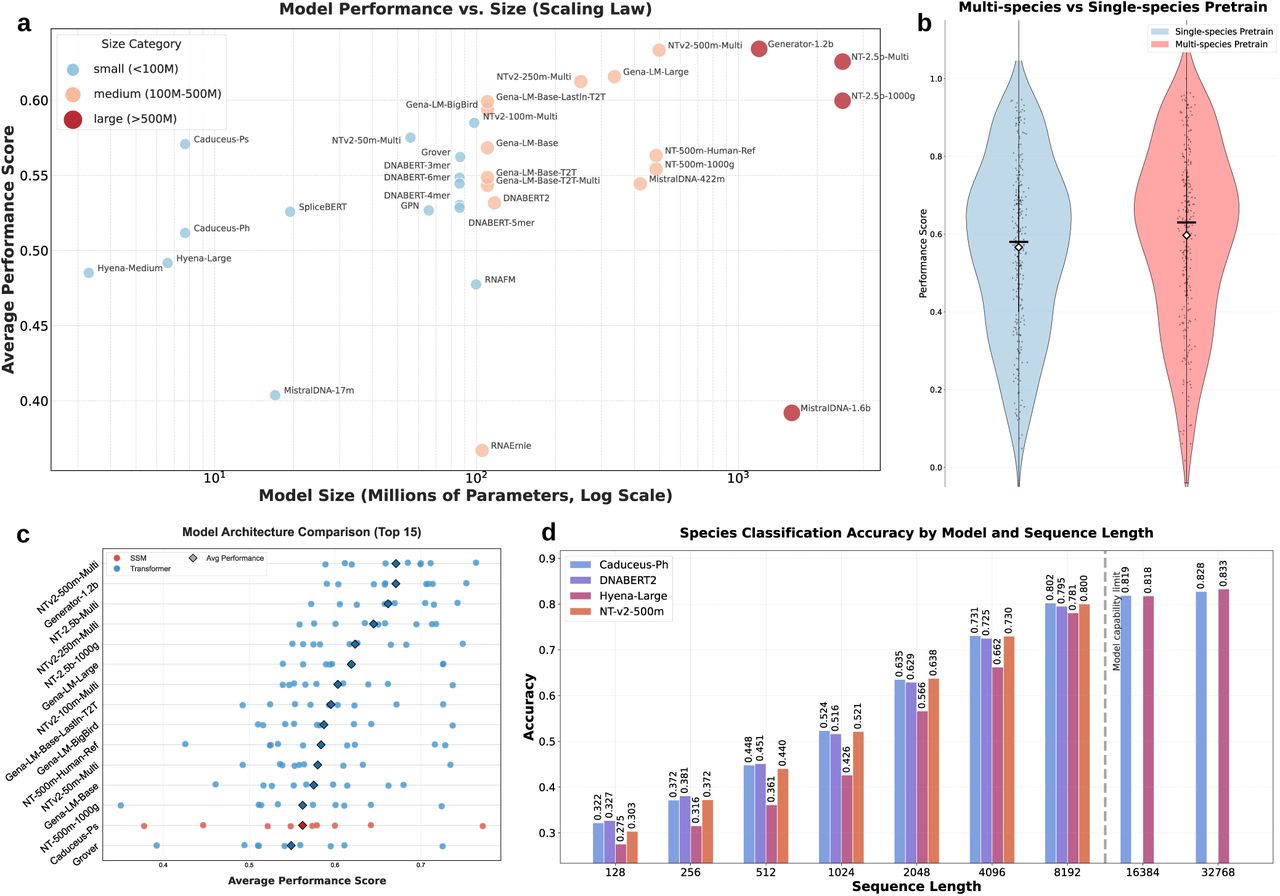
a:模型参数与平均基准测试性能的关系,每个点代表一个预训练模型,x轴为参数规模(对数尺度),y轴为平均评估分数;b:多物种预训练与单一人类数据预训练模型的性能分布对比;c:不同架构模型的性能对比,Transformer架构仍是主流,SSM-based模型具有潜力;d:输入序列长度达32768碱基对时的模型F1分数,长上下文窗口模型随序列长度增加增益显著。
4 讨论
4.1 核心结论
Genomic Touchstone建立了首个覆盖中心法则全链条的gLMs评估框架,证实了DNA预训练gLMs在跨模态任务中的泛化能力;Transformer模型整体表现最优,但高效架构在特定场景具有应用价值;模型性能受架构设计、缩放策略和预训练配置的共同影响,多物种、长序列预训练是关键优化方向。
4.2 研究局限与未来方向
- 局限包括:未开展从头预训练的对照实验、未涵盖生成式任务、以人类数据为中心、缺乏湿实验验证任务。
- 未来方向:开发兼顾性能与效率的新型架构、构建条件生成模型、扩展多物种评估框架、开发gLMs驱动的交互式AI实验辅助工具。
5 研究方法
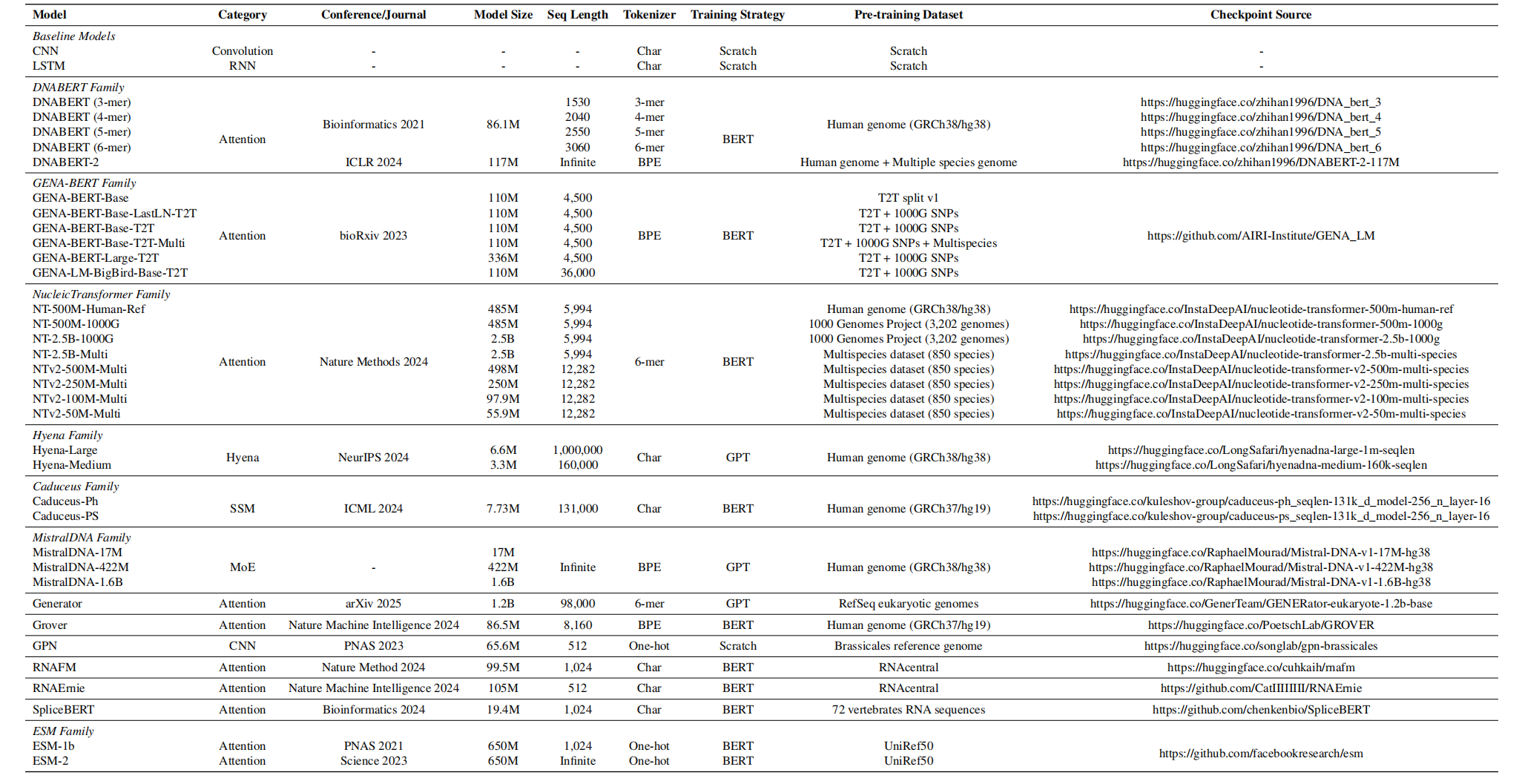
5.1 基准测试模型
涵盖基线模型(CNN、LSTM)、基因组语言模型(DNABERT系列、GPN、Nucleotide Transformer系列等)、RNA语言模型(RNAFM、SpliceBERT、RNAErnie)、蛋白质语言模型(ESM-1b、ESM-2),详细信息如下表:
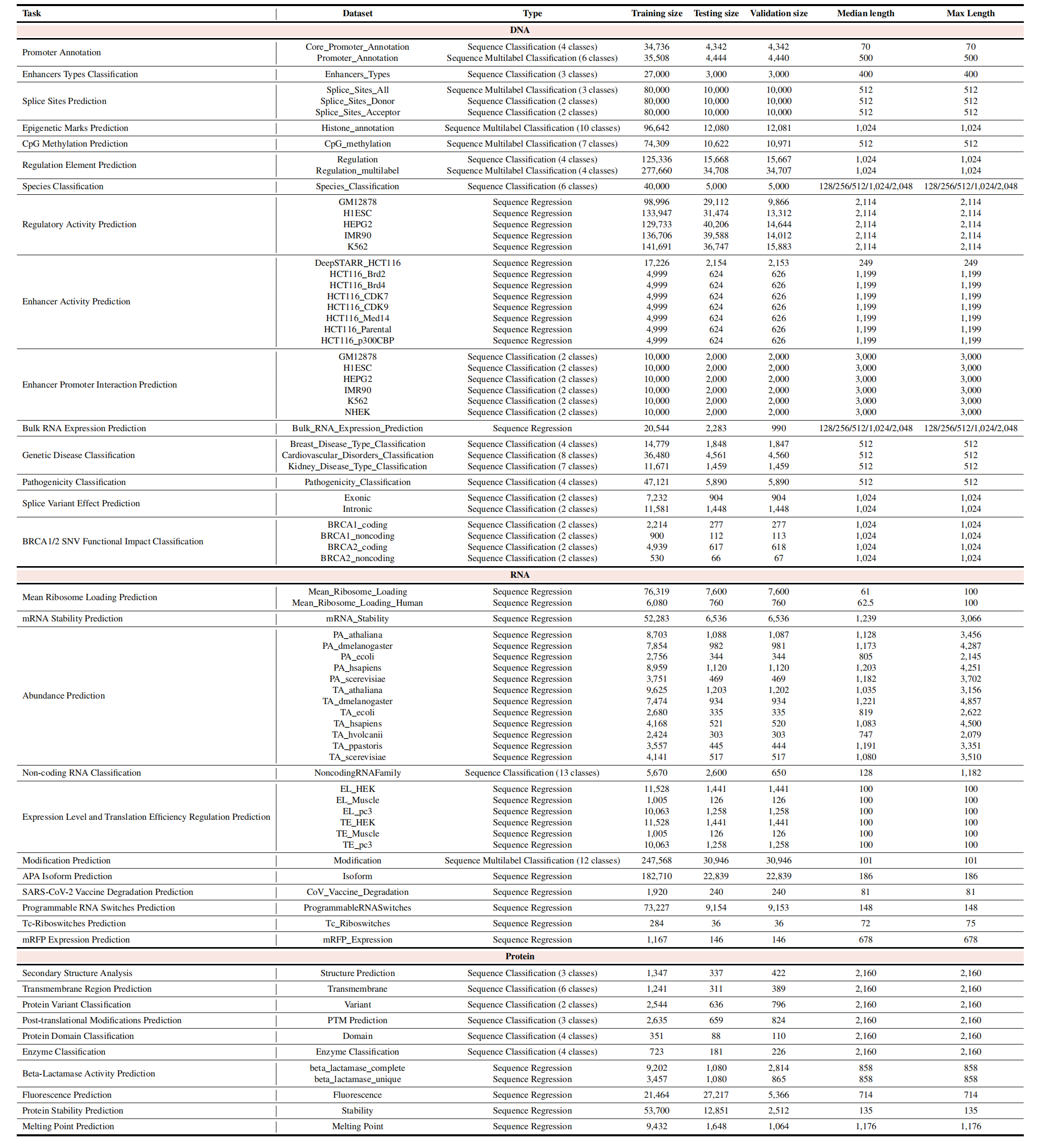
5.2 基准测试数据集
涵盖DNA、RNA、蛋白质三类模态的36项任务,88个数据集,关键信息如下(部分核心数据集):
5.3 实验设置
微调细节:所有模型使用预训练权重初始化,添加任务特定头(回归任务用单层回归头,分类任务用softmax分类头),AdamW优化器+余弦学习率调度器,基于验证集损失选择最优 epoch。
评估指标:
分类任务:准确率(Accuracy)、F1分数、宏F1分数(Macro-F1)
- $Accuracy = \frac{TP+TN}{TP+TN+FP+FN}$
- $F1 = 2 × \frac{Precision × Recall}{Precision + Recall}$(其中$Precision = \frac{TP}{TP+FP}$,$Recall = \frac{TP}{TP+FN}$)
- $Macro-F1 = \frac{1}{N} \sum_{i=1}^{N} F1_i$(N为类别数,$F1_i$为第i类的F1分数)
回归任务:决定系数($R^2$)、斯皮尔曼等级相关系数(ρ)
- $R^2 = 1 - \frac{\sum_{i=1}^{n}(y_i - \hat{y}i)^2}{\sum{i=1}^{n}(y_i - \overline{y})^2}$($y_i$为真实值,$\hat{y}_i$为预测值,$\overline{y}$为真实值均值)
- $\rho = 1 - \frac{6 \cdot \sum d_i^2}{n(n^2 - 1)}$($d_i$为预测值与真实值的秩差,n为样本数)
计算资源:5亿参数以下模型使用单张NVIDIA RTX 3090 GPU,5亿参数以上模型使用单张NVIDIA H800 GPU。
5.4 数据可用性
| Database | Link |
|---|---|
| DNA | |
| EPDnew | https://epd.expasy.org/epd/ |
| ENCODE | https://www.encodeproject.org/ |
| GENCODE | https://www.gencodegenes.org/ |
| Ensembl | https://www.ensembl.org |
| RefSeq | https://www.ncbi.nlm.nih.gov/refseq/ |
| GEO | https://www.ncbi.nlm.nih.gov/geo |
| ClinVar | https://www.ncbi.nlm.nih.gov/clinvar/ |
| SpliceVarDB | https://splicevardb.org |
| RNA | |
| Rfam | https://rfam.org |
| PaxDb | https://pax-db.org |
| GEO | https://www.ncbi.nlm.nih.gov/geo |
| RMBase | https://rna.sysu.edu.cn/rmbase |
| RADAR | http://rnaedit.com |
| EMBL-EBI | https://www.ebi.ac.uk/gxa/home |
| SRA | https://www.ncbi.nlm.nih.gov/sra |
| GenBank | https://www.ncbi.nlm.nih.gov/genbank |
| Ensembl | https://www.ensembl.org |
| Protein | |
| UniProt | https://www.uniprot.org/ |
| GENCODE | https://www.gencodegenes.org/ |
| Protein Data Bank | https://www.rcsb.org/ |
| ProteomicsDB | https://www.proteomicsdb.org/ |
参考
- Yihui Wang, Zhiyuan Cai, Qian Zeng, Yihang Gao, Jiarui Ouyang, Yingxue Xu, Shu Yang, Sunan He, Yuxiang Nie, Yu Cai, Fengtao Zhou, Cheng Jin, Xi Wang, Zhi Xie, Danqing Zhu, Ting Xie, Kwang-Ting Cheng, Can Yang, Xi Fu, Jiguang Wang, Kang Zhang, Jianhua Yao, Raul Rabadan, Hao Chen. Genomic Touchstone: Benchmarking Genomic Language Models in the Context of the Central Dogma. bioRxiv 2025.06.25.661622; doi: https://doi.org/10.1101/2025.06.25.661622
加关注
关注公众号“生信之巅”。
 |
 |



