介绍
DeepARG 是一种机器学习解决方案,它使用深度学习来表征和注释宏基因组中的抗生素抗性基因。它由两种输入模型组成:短序列Reads和gene-like序列。
安装软件
通过
conda安装1
2
3
4
5
6
7
8
9
10
11
12
13
14
15# 创建环境
conda create -n deeparg_env python=2.7.18
conda activate deeparg_env
# 安装diamond
conda install -c bioconda diamond==0.9.24
# 安装其他依赖
conda install trimmomatic vsearch bedtools bowtie2 samtools
# 安装DeepARG
pip install deeparg==1.0.2
# 下载数据库等, -o指定下载路径
deeparg download_data -o tools/deeparg通过其他方法安装
参考官方文档进行。
使用软件
预测Reads中的ARGs
输入文件
双端Reads。
命令
1 | deeparg short_reads_pipeline --forward_pe_file Reads/LD201221-0003_S20210104-0015_F01_clean.R1.fq.gz --reverse_pe_file Reads/LD201221-0003_S20210104-0015_F01_clean.R2.fq.gz --output_file F01.deeparg -d ~/tools/deeparg/ |
+++primary 参数解析
-h, --help show this help message and exit--forward_pe_file FORWARD_PE_FILE: forward mate from paired end library--reverse_pe_file REVERSE_PE_FILE: reverse mate from paired end library--output_file OUTPUT_FILE: save results to this file prefix-d DEEPARG_DATA_PATH: Path where data was downloaded [see deeparg download-data –help for details]--deeparg_identity DEEPARG_IDENTITY: minimum identity for ARG alignments [default 80]--deeparg_probability DEEPARG_PROBABILITY: minimum probability for considering a reads as ARG-like [default 0.8]--deeparg_evalue DEEPARG_EVALUE: minimum e-value for ARG alignments [default 1e-10]--gene_coverage GENE_COVERAGE: minimum coverage required for considering a full gene in percentage. This parameter looks at the full gene and all hits that align to the gene. If the overlap of all hits is below the threshold the gene is discarded. Use with caution [default 1]
+++
预测FASTA序列中的ARGs
输入文件
可以是核苷酸序列或者是氨基酸序列。
命令
1 | # 1) Annotate gene-like sequences when the input is a nucleotide FASTA file: |
+++primary 参数解析
usage: deeparg predict
-h, --help show this help message and exit
--model MODEL: Select model to use (short sequences for reads | long sequences for genes) SS|LS [No default]
-i, --input-file INPUT_FILE: Input file (Fasta input file)
-o, --output-file OUTPUT_FILE: Output file where to store results
-d, --data-path DATA_PATH: Path where data was downloaded [see deeparg download-data –help for details]
--type TYPE: Molecular data type prot/nucl [Default: nucl]
--min-prob MIN_PROB: Minimum probability cutoff [Default: 0.8]
--arg-alignment-identity ARG_ALIGNMENT_IDENTITY: Identity cutoff for sequence alignment [Default: 50]
--arg-alignment-evalue ARG_ALIGNMENT_EVALUE: Evalue cutoff [Default: 1e-10]
--arg-alignment-overlap ARG_ALIGNMENT_OVERLAP: Alignment read overlap [Default: 0.8]
--arg-num-alignments-per-entry ARG_NUM_ALIGNMENTS_PER_ENTRY: Diamond, minimum number of alignments per entry [Default: 1000]
--model-version MODEL_VERSION: Model deepARG version [Default: v2]
+++
输出
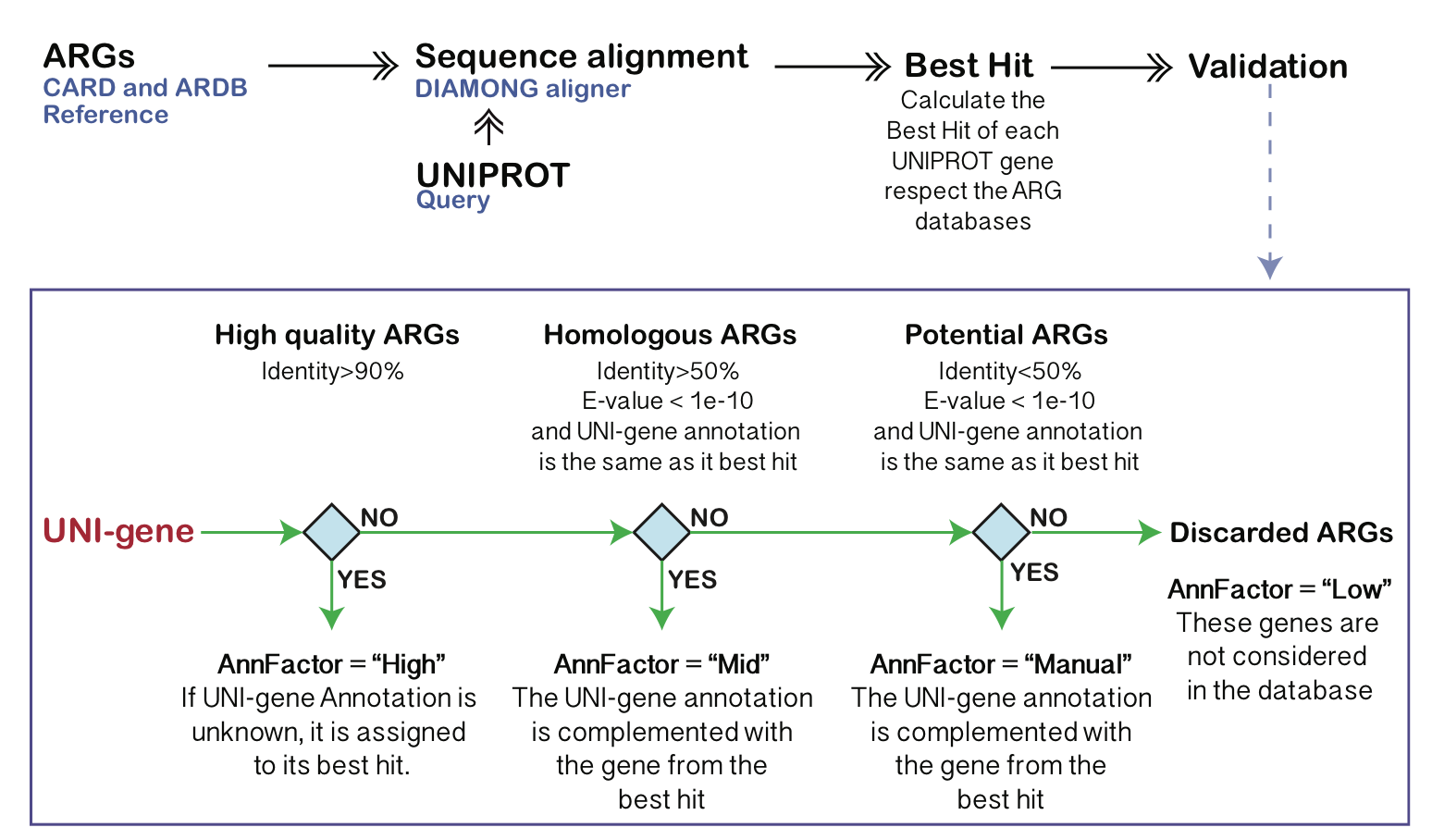
DeepARG产生两个文件: .ARG包含probability >= –prob (0.8 default)的序列,.potential.ARG包含probability < –prob (0.8 default)的序列。*.potential.ARG文件中仍可能含有ARG-like序列,但是需要对这些序列进行检查。
两个文件的文件头如下所示:
1 | * ARG_NAME |
批处理
- Run_deepARG_reads.pl
- Run_deepARG_scafs.pl
样本间比较
生成样本VS. ARGs矩阵
- co_type.pl
绘制气泡图
- Bubble_plot.R
绘制热图
- Plot_heatmap.R
参考
代码获取
关注公众号“生信之巅”,聊天窗口回复“92eb”获取下载链接。
 |
 |
敬告:使用文中脚本请引用本文网址,请尊重本人的劳动成果,谢谢!