
下载有summary的基因组
在NCBI基因组数据库搜索物种
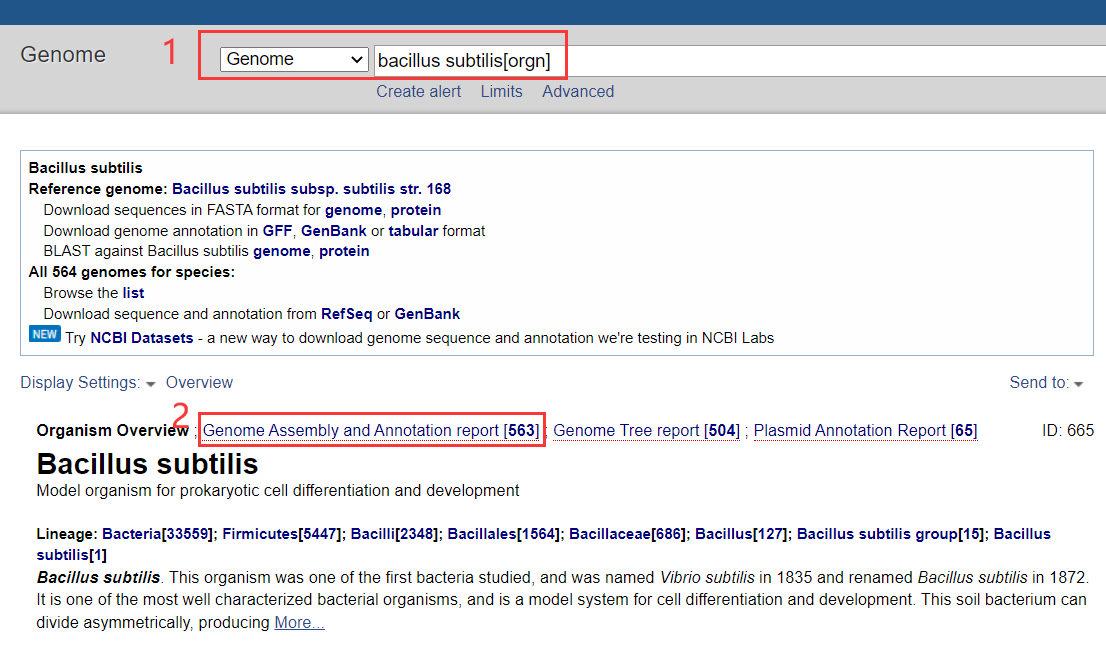
下载元数据
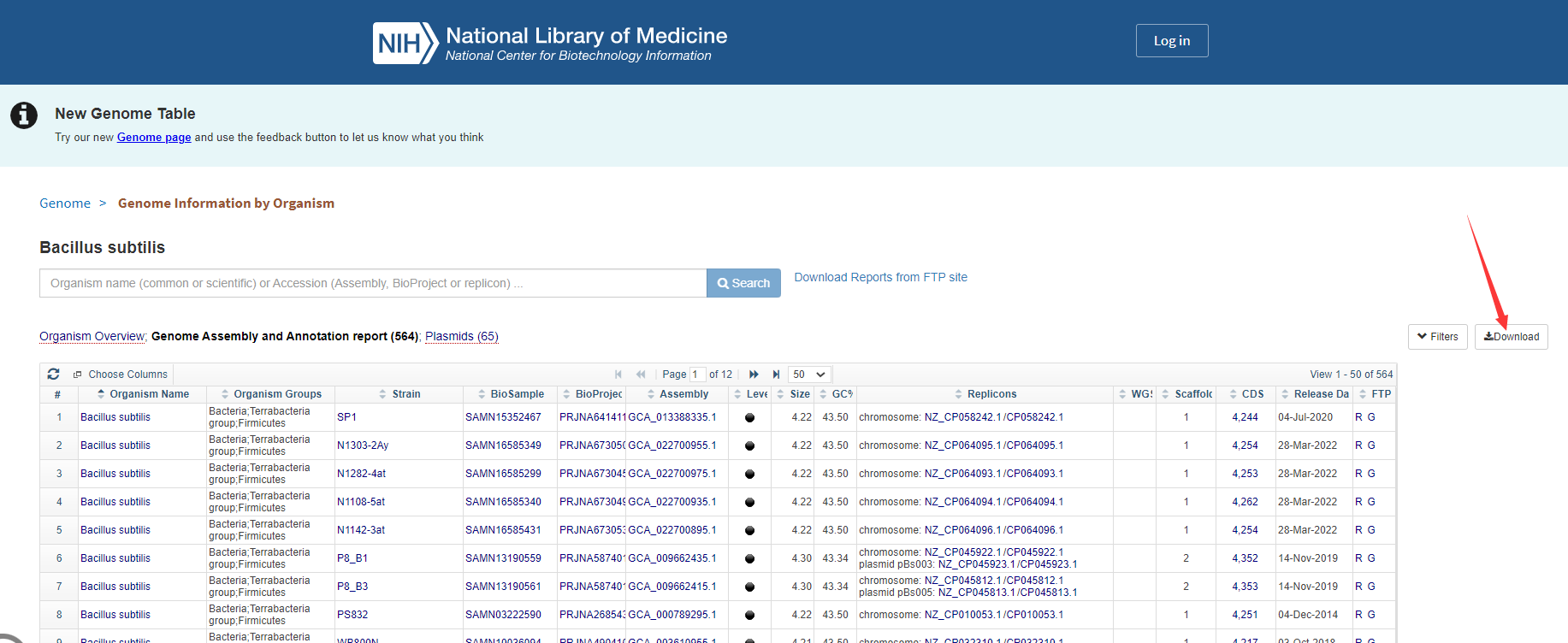
获取下载链接
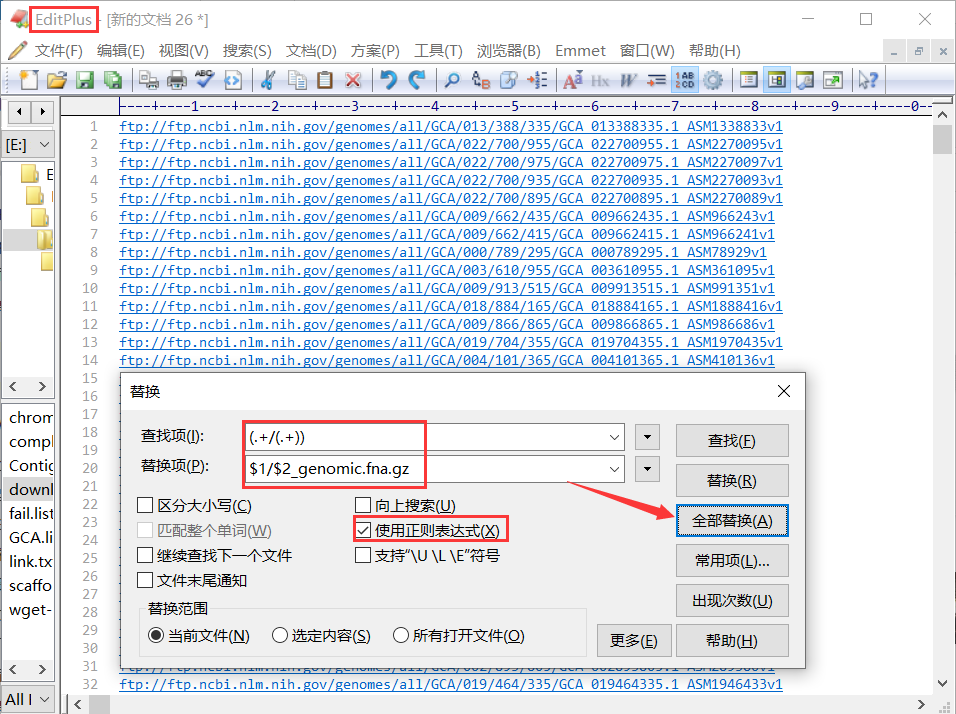
打开下载的元数据文件prokaryotes.csv(该文件也可以直接去NCBI FTP中下载,一般在各物种的目录下,名字为assembly_summary.txt,其格式与prokaryotes.csv略有不同,但都含有链接),将倒数第二列或最后一列的链接拷贝到TXT文本文档中,在每一行的最后加上要下载的文件名和数据类型:- 基因组:文件名 _genomic.fna.gz
- 蛋白序列:文件名 _protein.faa.gz
- CDs序列:文件名 _cds_from_genomic.fna.gz
- …
可参考下图中的示例进行命名:
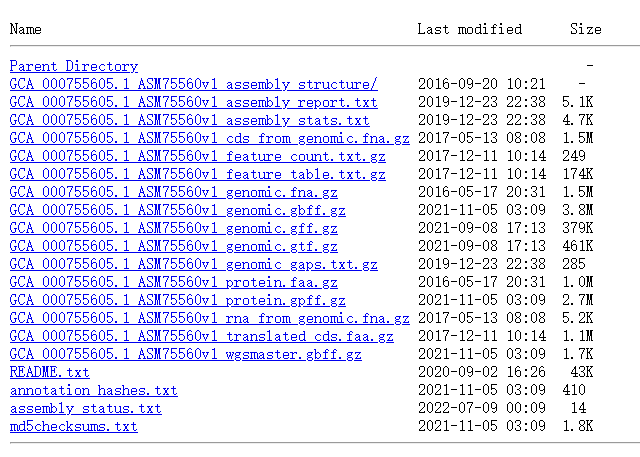
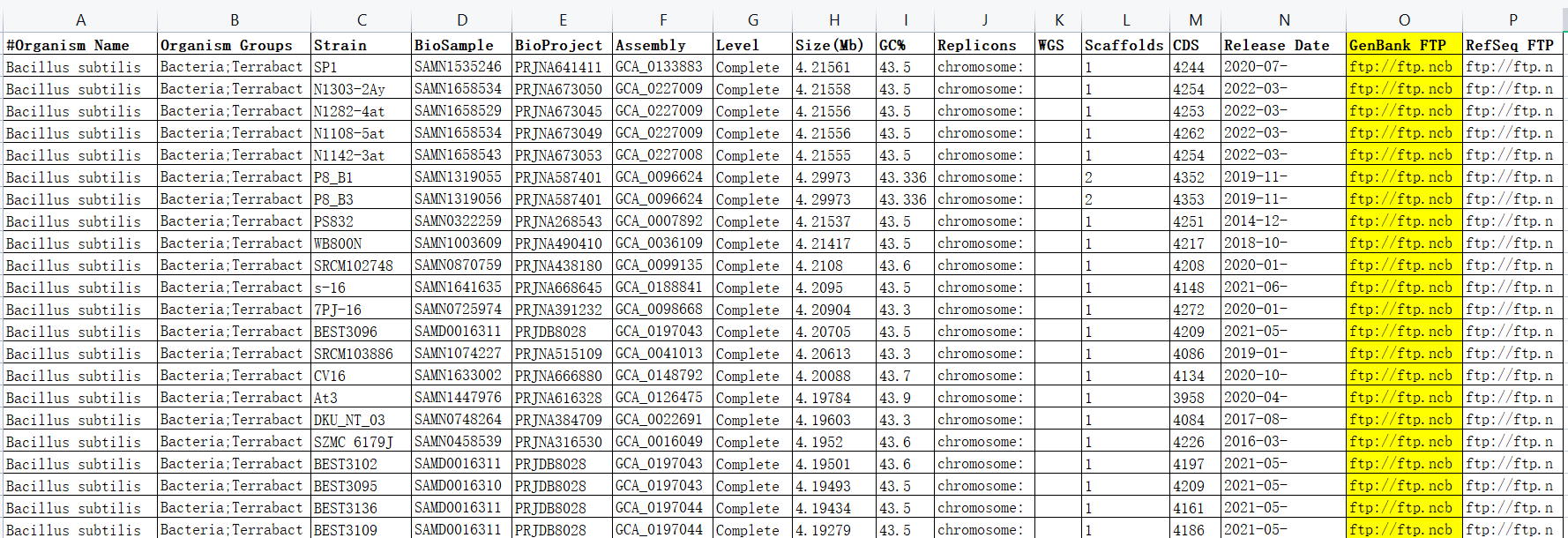
建议用正则表达式替换(依赖EditPlus或其他具有正则表达式功能的文本编辑器),以基因组序列为例:
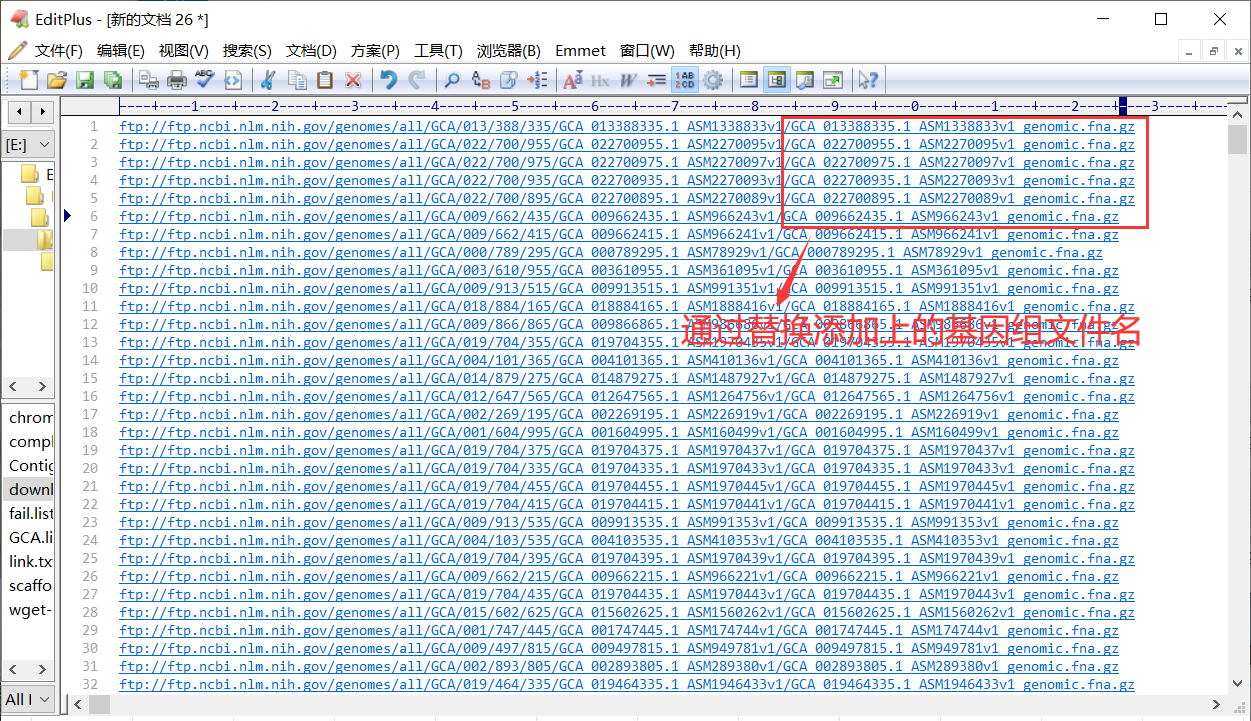
开始下载
将获得的完整链接保存到名字为link.txt的文本文档中,在LINUX终端中运行for link in $(cat link.txt); do wget -c $link; done即可批量下载基因组到genome目录中。若不慎在行末引入了看不见的换行符,可以用命令perl -pe 's/[\n\r]+//g' link.txt > link2.txt进行删除。再用for link in $(cat link2.txt); do wget -c $link; done下载。
SRA数据下载
安装SRA Toolkit(建议用方式2安装)
方式1: 手动安装(需要单独安装parallel,见方式2)
-
根据自己的系统选择合适的版本进行下载,并将软件包中
bin的绝对路径或相对路径加入到环境变量中,以便可以在终端中直接调用。 设置默认下载目录
如果不设置,默认会将基因组下载到家目录下,通过在终端里输入
vdb-config -i命令配置下载目录,即下载到当前目录下。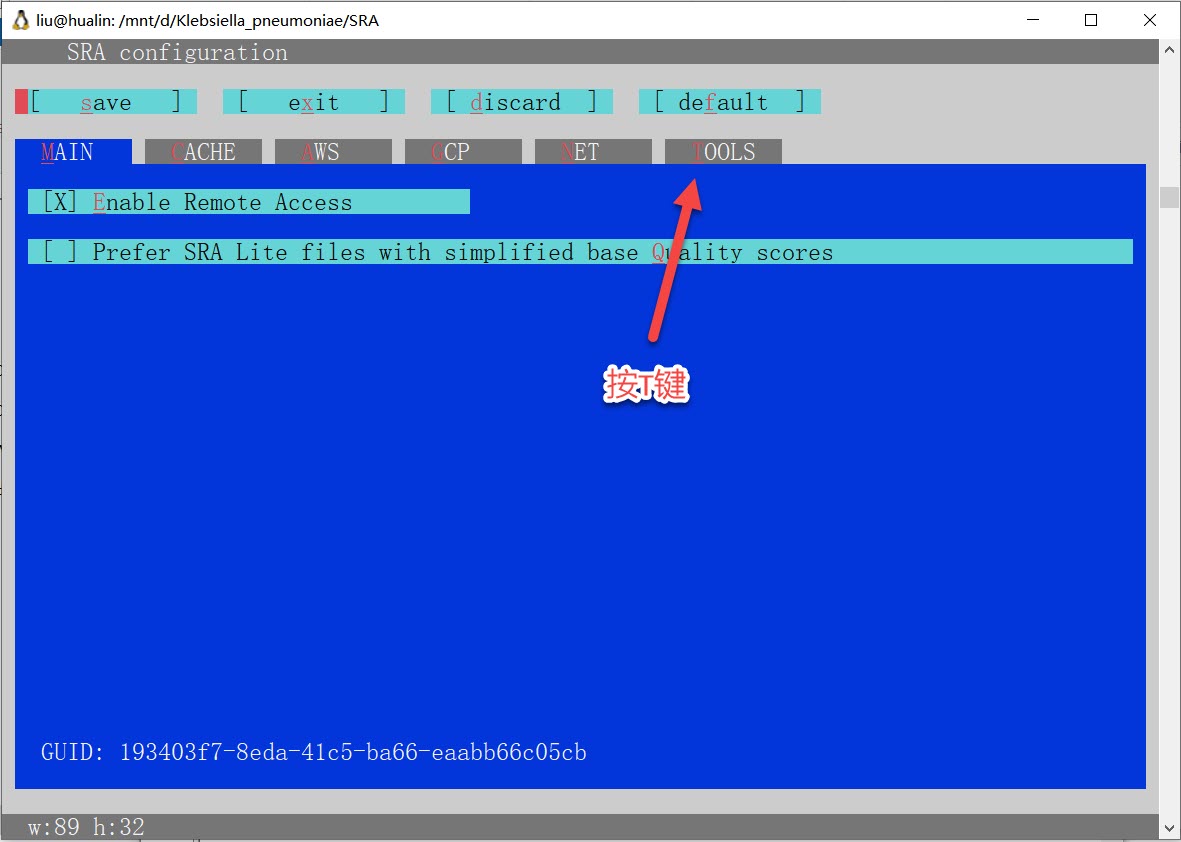
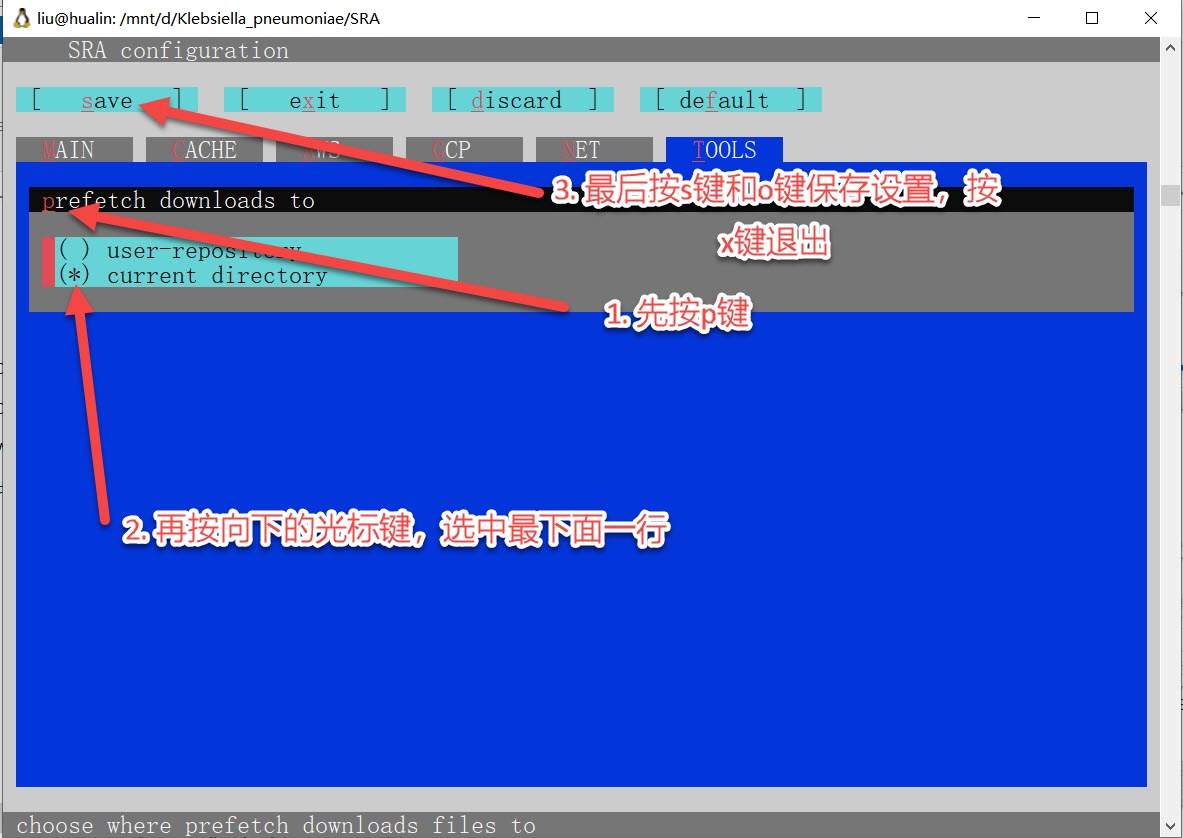
方式2: 通过conda安装
安装SRA Toolkit
1
mamba install -c bioconda sra-tools
安装parallel
用于多线程下载,可提高下载速度。
1
mamba install -c conda-forge parallel
准备SRA号列表文件
准备包含
SRA号的列表文件,每一行含有一个编号,文件命名为srr_list.txt,如下所示:1
2
3
4
5
6
7
8DRR634073
DRR634074
DRR634118
DRR634144
DRR634148
DRR634091
DRR634127
DRR634140
开始下载
将列表文件与脚本downloadSRA.sh放在同一目录下,在Linux终端中运行bash downloadSRA.sh,下载的文件存放于fastq_output目录中。
1 | bash downloadSRA.sh |
利用FTP软件根据物种下载基因组
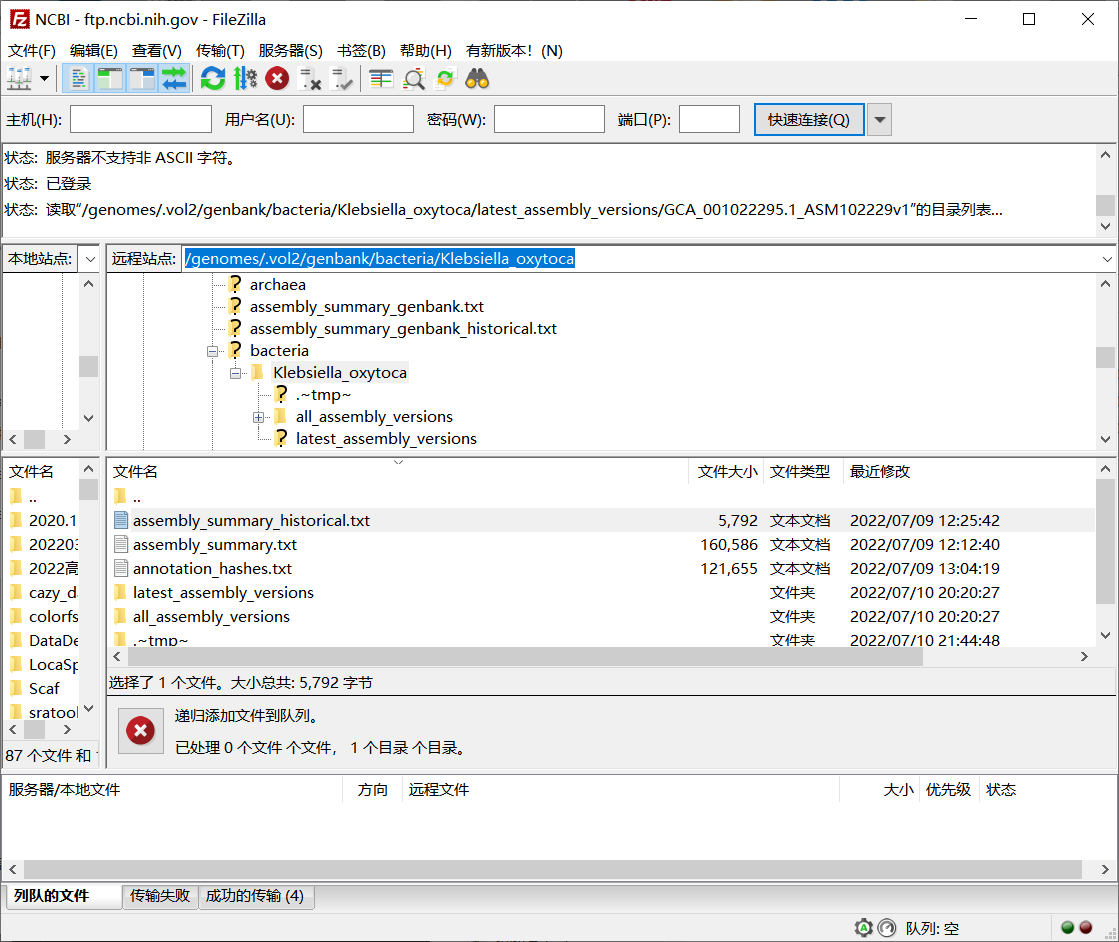
在右侧的列表中选中所有目录拖拽到本地即可开始下载。
根据WGA assession number下载基因组
-
1
conda install -c bioconda ncbi-genome-download
准备WGA assession number列表文件
开始下载
终端里输入如下命令:1
ncbi-genome-download --assembly-accessions GCA.txt --output-folder 6_3 bacteria --section genbank --formats fasta
其中
GCA.txt为包含assession number的列表文件,每行一个assession number。
该软件的问题在于,需要科学上网,所以很多时候会掉链子。
代码获取
关注公众号“生信之巅”,聊天窗口回复“213”获取下载链接。
 |
 |
敬告:使用文中脚本请引用本文网址,请尊重本人的劳动成果,谢谢!



