
安装软件
1 | mamba install miniasm minipolish raven-assembler flye medaka python=3.8 fastp bwa masurca trycycler |
Step 1: Generating assemblies
准备三代数据
如果是含有多个文件的话,需要合并到一个文件里
1 | cat WHQ17/barcode14/*.fastq > WHQ17.fastq |
质控
This will discard short reads (less than 1 kbp) and very bad reads (the worst 5%)
1 | filtlong --min_length 1000 --keep_percent 95 WHQ17.fastq > WHQ17.good.fastq |
取子集 Subsampling reads
1 | trycycler subsample --reads WHQ17.good.fastq --out_dir read_subsets --threads 8 |
+++ info 参数解析
- --count: 输出的Reads子集的数量,大部分情况使用默认的12即可。
- --genome_size: 预估的基因组大小 (e.g. –genome_size 5.5m)。不提供的话会通过miniasm 组装基因组,以估计大小,但是速度会慢。此值用于计算reads深度,不需要很精确,10%的错误是允许的。
- --min_read_depth: 允许的最小read深度,控制着取子集的深度。
- --threads: 使用的线程数(越大越好),影响miniasm 的组装速度。
+++
得到:read_subsets/sample_*.fastq
组装 Generating assemblies
1 | threads=6 # change as appropriate for your system |
得到:assemblies/*.fasta
删除中间文件:
1 | rm -r read_subsets |
Step 2: Clustering contigs
Running Trycycler cluster
1 | trycycler cluster --assemblies assemblies/*.fasta --reads WHQ17.good.fastq --out_dir trycycler --threads 10 |
+++ info 参数解析
- --min_contig_len: 最小contig长度(默认1000),短于该长度的contigs将被丢弃。如果你的基因组含有长度更小的质粒,将该值设低。
- --min_contig_depth: 覆盖contigs的reads的最低深度。For example, if an assembly has a mean depth of 90× and this setting is 0.1 (the default), then any contig with <9× depth will be removed.
- --distance: this is the Mash distance threshold used when defining clusters, and the default threshold is 0.01. Smaller thresholds (e.g. 0.005) can result in a larger number of tighter clusters. Larger thresholds (e.g. 0.02) can result in a smaller number of looser clusters.
- --threads: this is how many threads Trycycler will use for read alignment. It will only affect the speed performance, so you’ll probably want to use as many threads as you have available.
+++
输出
trycycler/contigs.phylip: a matrix of the Mash distances between all contigs in PHYLIP format.trycycler/contigs.newick: a FastME tree of the contigs built from the distance matrix. This can be visualised in a phylogenetic tree viewer such as FigTree, Dendroscope or Archaeopteryx.- One directory for each of the clusters:
trycycler/cluster_001, trycycler/cluster_002, etc. These directories will each contain a subdirectory named 1_contigs which includes the FASTA files for the contigs in the cluster.
筛选 clusters
将contigs.newick导入进化树查看软件,肉眼观察,主观意愿挑选,有问题的cluster删掉。参照Trycycler官方说明。
Step 3: Reconciling contigs
Running Trycycler reconcile
1 | trycycler reconcile --reads WHQ17.good.fastq --cluster_dir trycycler/cluster_001 --threads 10 |
+++ info 参数解析
General settings:
- --linear: use this option if your input contigs are not circular. It will disable the circularisation-correction steps in Trycycler reconcile.
- --threads: this is how many threads Trycycler will use for read alignment. It will only affect the speed performance, so you’ll probably want to use as many threads as you have available.
- --verbose: use this flag to display extra output. Mainly there for debugging purposes.
Initial check:
- --max_mash_dist: if any of the sequences have a pairwise Mash distance of more than this (default = 0.02), then the contigs will fail the initial check.
- --max_length_diff: if any of the sequences have a pairwise relative length factor of more than this, then the contigs will fail the initial check. For example, if set to 1.1 (the default), then no contig can be more than 10% longer than any other.
Circularisation:
- --max_add_seq and –max_add_seq_percent: these control how much sequence Trycycler is willing to add to a contig to circularise it. For example, if they are set to 1000 and 5 (the defaults), then Trycycler will be willing to add up to 1000 bp or 5% of a contig’s length (whichever is smaller) to circularise it. Any contig which requires more than 1000 bp or 5% of its length added to circularise will cause Trycycler reconcile to fail.
- --max_trim_seq and –max_trim_seq_percent: these control how much sequence Trycycler is willing to remove from a contig to circularise it. For example, if they are set to 50000 and 10 (the defaults), then Trycycler will be willing to remove up to 50000 bp or 10% of a contig’s length (whichever is smaller) to circularise it. Any contig which requires more than 50000 bp or 10% of its length removed to circularise will cause Trycycler reconcile to fail.
Final check:
- --min_identity: if any of the sequences have a pairwise global alignment percent identity of less than this (default = 98), then the contigs will fail the final check.
- --max_indel_size: if any of the sequences have a pairwise alignment indel size of more than this (default = 250), then the contigs will fail the final check.
+++
输出
When finished, Trycycler reconcile will make 2_all_seqs.fasta in the cluster directory, a multi-FASTA file containing each of the contigs ready for multiple sequence alignment.
Dotplots
You can optionally run Trycycler dotplot on any problematic clusters to visualise the relationship between the sequences. For example:
1 | trycycler dotplot --cluster_dir trycycler/cluster_002 |
This will create an image file (dotplots.png) in the cluster directory with a dotplot for all pairwise combinations of sequences. Same-strand k-mer matches are drawn in blue, and opposite-strand k-mer matches are drawn in red. For example:
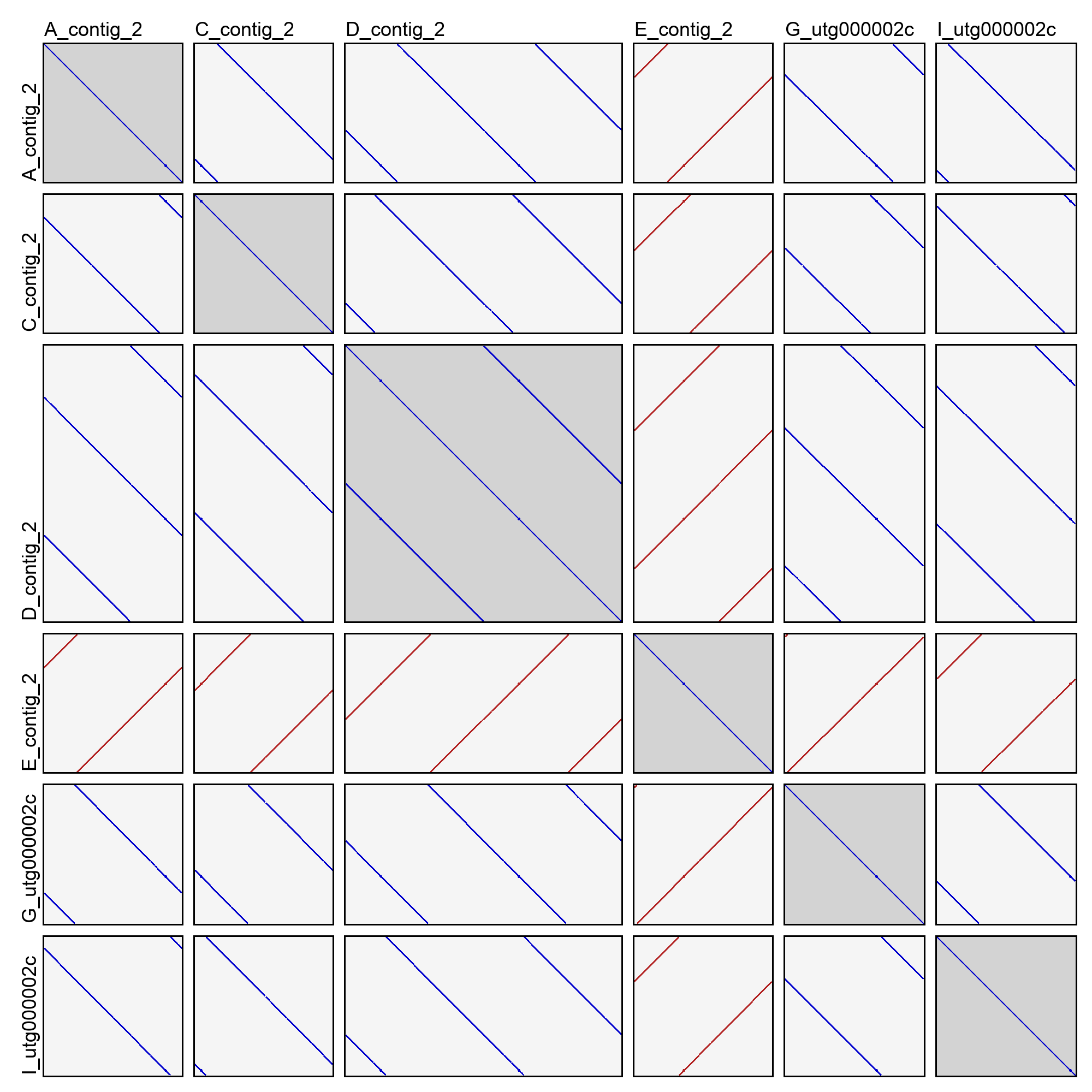
In the above example (taken from cluster 2 of the good demo dataset), you can see that most of the sequences are in very nice agreement with each other. They have shifted start positions relative to each other, but that’s expected for contigs of a circular sequence. One of the contigs (E_contig_2) is on the opposite strand as the others, but that too is normal. Sequence D_contig_2, however, is a problem! It contains two entire copies of the same sequence, visible in the dotplot with itself and the dotplots with other sequences. It will need to be excluded or trimmed in order for reconciliation to succeed.
Step 4: Multiple sequence alignment
Running Trycycler msa
1 | trycycler msa --cluster_dir trycycler/cluster_001 --threads 8 |
+++ info 参数解析
除了线程数外,其他三个参数可以用默认值。
- --kmer: the k-mer size used for sequence partitioning (default = 32).
- --step: the step size used for sequence partitioning (default = 1000).
- --lookahead: the look-ahead margin used for sequence partitioning (default = 10000).
- --threads: this is how many parallel instances of MUSCLE will be used when aligning the sequence partitions. It will only affect the speed performance, so you’ll probably want to use as many threads as you have available.
+++
输出
When finished, Trycycler reconcile will make a 3_msa.fasta file in the cluster directory, a FASTA-formatted multiple sequence alignment of the contigs ready for use in generating a consensus.
Step 5: Partitioning reads
Running Trycycler partition
1 | trycycler partition --reads WHQ17.good.fastq --cluster_dirs trycycler/cluster_* --threads 8 |
+++ info 参数解析
- --min_aligned_len: reads with less than this many bases aligned (default = 1000) will be ignored.
- --min_read_cov: reads with less than this percentage of their length covered by alignments (default = 90.0) will be ignored.
- --threads: this is how many threads Trycycler will use for read alignment. It will only affect the speed performance, so you’ll probably want to use as many threads as you have available.
+++
输出
After Trycycler partition completes, each of the cluster directories should have a 4_reads.fastq file which contains its share of the total reads.
Step 6: Generating a consensus
Running Trycycler consensus
1 | trycycler consensus --cluster_dir trycycler/cluster_001 --threads 8 |
+++ info 参数解析
- --linear: use this option if your input contigs are not circular. It will disable the circularisation steps when aligning reads and choosing variants.
- --min_aligned_len: reads with less than this many bases aligned (default = 1000) will be ignored.
- --min_read_cov: reads with less than this percentage of their length aligned (default = 90.0) will be ignored.
- --threads: this is how many threads Trycycler will use for read alignment. It will only affect the speed performance, so you’ll probably want to use as many threads as you have available.
- --verbose: use this flag to display extra output. For every read-assessed variant, this will show the alternative sequences and their read alignment scores.
+++
输出
When finished, you should have a 7_final_consensus.fasta file in each of your cluster directories. If you have multiple clusters, you can combine their consensus sequences into a single FASTA file like this:
1 | cat trycycler/cluster_*/7_final_consensus.fasta > trycycler/consensus.fasta |
Step 7: Polishing after Trycycler
Medaka (需要知道测序仪信息basecalling)
1 | for c in trycycler/cluster_*; do |
1 | cat trycycler/cluster_*/8_medaka.fasta > trycycler/consensus.fasta |
Short-read polishing
Step 1: read QC
1 | fastp --in1 WHQ17_BDMS190038054-1a_1.clean.fq.gz --in2 WHQ17_BDMS190038054-1a_2.clean.fq.gz --out1 WHQ17_1.fastq.gz --out2 WHQ17_2.fastq.gz --unpaired1 WHQ17_u.fastq.gz --unpaired2 WHQ17-u.fastq.gz |
Step 2: Polypolish
1 | bwa index trycycler/consensus.fasta |
Step 3: POLCA
1 | # 这一步报错,暂未执行 |
删除无用信息
1 | rm alignments_1.sam alignments_2.sam fastp.* |



