寻找同源蛋白家族用的比较多的是Orthomcl,但是该软件多年前已经停止更新,且使用的时候需要安装和使用MySQL,操作起来比较繁琐。因此OrthoFinder应运而生,并且更新到版本2。后者不但可以寻找同源家族,并且可以构建基因家族进化树。
- OrthoFinder及依赖包的安装
- 下载
1
wget https://github.com/davidemms/OrthoFinder/releases/download/v2.2.7/OrthoFinder-2.2.7.tar.gz
- 解压
1
tar zxvf OrthoFinder-2.2.7.tar.gz
- 安装(加入环境变量即可)依赖包
1
2
3
4
5
6
7
8
9
10
11
12
13vim ~/.bashrc
i
export PATH=$PATH:$HOME/tools/OrthoFinder-2.2.7
Esc
shift + ;
wq!
source ~/.bashrc
(1)DIAMOND
下载对应版本,解压并将主程序拷贝至存在于环境变量的目录下或将其所在的目录加入环境变量:
1 | wget https://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz |
没有root权限的可以把diamond所在目录加入环境变量。
(2) MMseqs2
下载对应版本,解压并将主程序拷贝至存在于环境变量的目录下或将其所在的目录加入环境变量:
1 | wget https://github.com/soedinglab/MMseqs2/releases/download/7-4e23d/MMseqs2-Linux-AVX2.tar.gz |
(3) MCL
Ubuntu, Debian, Linux Mint安装方法:
1
sudo apt-get install mcl
Centos, Redhat安装方法:
1
2
3
4
5
6
7wget https://micans.org/mcl/src/mcl-latest.tar.gz
tar zxvf mcl-latest.tar.gz
cd mcl-14-137(视具体情况而定)
./configure
make
make check
sudo make install(4) FastME
下载二进制文件,解压并将主程序拷贝至存在于环境变量的目录下或将其所在的目录加入环境变量:
1 | wget http://www.atgc-montpellier.fr/download/sources/fastme/fastme-2.1.5.tar.gz |
(5) 可选: BLAST+
- Ubuntu, Debian, Linux Mint安装方法:
1
sudo apt-get install ncbi-blast+
- Centos, Redhat安装方法:
1
2
3
4
5
6
7
8
9wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.8.1+-x64-linux.tar.gz
tar zxvf ncbi-blast-2.8.1+-x64-linux.tar.gz
vim ~/.bashrc
i
export PATH=$PATH:$HOME/tools/ncbi-blast-2.8.1+/bin
Esc
shift + ;
wq!
source ~/.bashrc
- 运行 OrthoFinder
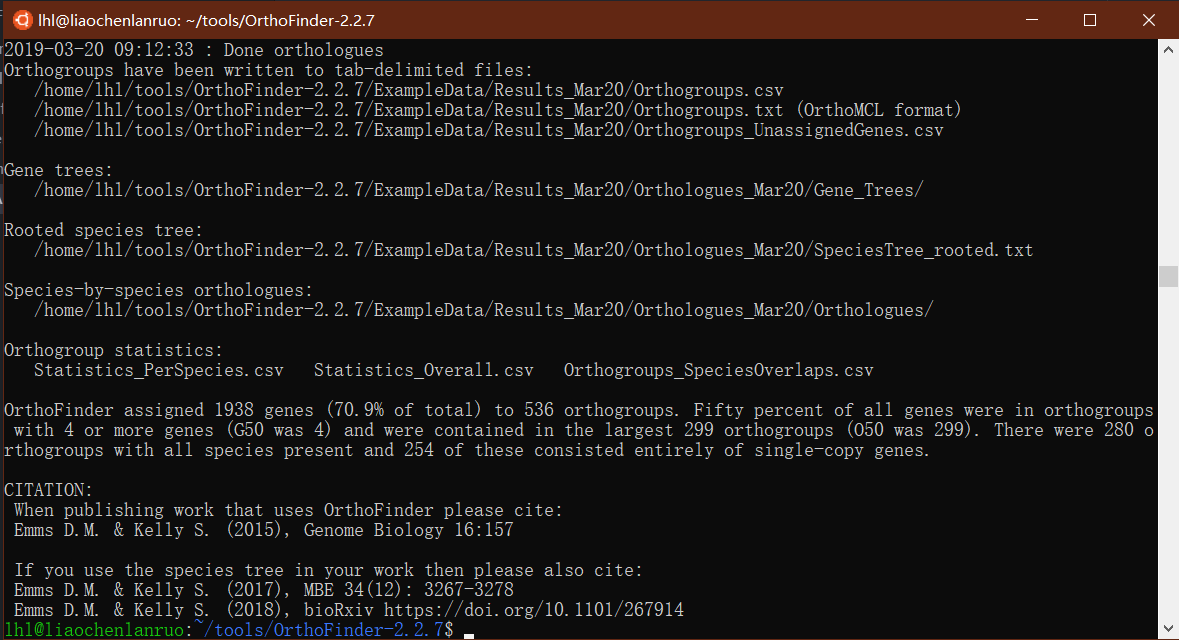
(1) 运行示例数据:运行结果如下,会显示输出文件的路径,表明运行成功:1
2cd OrthoFinder-2.2.7
orthofinder -f ExampleData
(2) 运行自己的数据:
- step1:数据准备
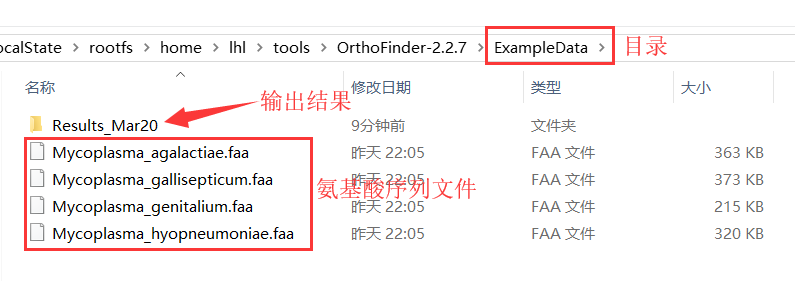
下载氨基酸序列,要求为fasta格式,每个物种一个文件。将所有fasta文件存于一个目录中(如Data目录),如下图所示。文件名要简洁并有区分性,因为文件名会作为最终的物种ID。
- step2:运行程序
在Data目录的上一级目录打开终端,运行如下命令:
1 | orthofinder -f Data -t 线程数 |
- 结果解读
(1) Results Files: Orthogroups
包含一个主文件“Orthogroups.csv”和两个支持文件:
Orthogroups.csv,每一行为一个group,每一列为一个物种,行列交汇处为基因名称。
Orthogroups_UnassignedGenes.csv,包含所有未分配到任何group的基因名称。
Orthogroups.txt,OrthoMCL格式的输出结果,内容等同于Orthogroups.csv。
(2)Results Files: Orthogroup Statistics
包含一些统计数据,可用于比较基因组分析、绘图以及质控。
Statistics_Overall.csv和Statistics_PerSpecies.csv,提供基本的描述信息
Orthogroups_SpeciesOverlaps.csv,两两物种的group共享矩阵
Species-specific orthogroup:该group仅包含一个物种的基因。
G50:group中的基因数,使得50%的基因处于该大小或更大的group中。
O50:最小数量的group,使得50%的基因处于该大小或更大的group中。
Single-copy orthogroup:每个物种中只有一个基因的group(相当于单拷贝核心基因)。这些group是构建物种树和许多其他分析的理想选择。
Unassigned gene:未与任何其他基因划分到一个group的基因。
(3) Results Files: Orthologues
两两物种间的直系同源基因,每一行为一个group,第一列为group编号,第二列为第一个物种的基因,第三列为第二个物种的基因。同一物种的基因名以“,”分割。直向同源物可以是一对一,一对多或多对多。
(4) Results Files: Gene Trees and Species Tree
每个group的基因树和定根的物种树以newick格式输出,可以用各种看树软件展示,如MEGA、iTOL、Dendroscope和FigTree等,个人推荐用iTOL。
- 高级用法
(1)添加新物种到之前的分析
(previous_orthofinder_directory指的是包含“SpeciesIDs.txt”的目录)
1 | orthofinder -b previous_orthofinder_directory -f new_fasta_directory |
(2)从之前的分析中移除物种
从输出目录下找到工作目录“WorkingDirectory”中的“SpeciesIDs.txt”文件,在要移除的物种那一行最前面加上一个“#”并保存,然后运行(previous_orthofinder_directory指的是包含“SpeciesIDs.txt”的目录):
1 | orthofinder -b previous_orthofinder_directory |
(3)同时添加和删除物种
编辑好“SpeciesIDs.txt”后,运行:
1 | orthofinder -b previous_orthofinder_directory -f new_fasta_directory |
(4)更多高级功能请阅读官方文档
主要包括“Inferring MSA Gene Trees”、并行计算、单独运行BLAST、使用预先计算的BLAST结果以及回归检测。